Veritaudit
Veritaudit
Näillä sivuilla esitetään kansalliset veritautien diagnoosi-, seuranta- ja hoito-ohjeet, joita on tuotettu Suomen Leukemiaryhmän tautikohtaisten työryhmien toimesta. Ohjeiston sisällön omistaja on Suomen Hematologiyhdistys.
Akuutit leukemiat
Akuutit leukemiat
ALL
ALL
| Kirjoittajat: | Erkki Elonen, Kimmo Porkka, Ulla Wartiovaara-Kautto |
| Viimeisin päivitys: | 8.10.2014 |
| ©Suomen Hematologiyhdistys | Suomen Leukemiaryhmä |
Diagnoosi
Diagnoosi
ALL-taudin diagnoosi perustuu luuytimen (tai veren) morfologiseen tutkimukseen, sekä blastien virtaussytometriseen pintaproteiinitutkimukseen, joita täydennetään myöhemmin valmistuvilla sytomolekyyligeneettisillä tutkimuksilla.
Diagnostiset kriteerit (WHO2008): blastien määrä veressä ja/tai luuytimessä ≥25% (poikkeus: tyyppitranslokaatiot).
Diagnoosivaiheen tutkimusten tulosten perusteella arvioidaan potilaan hoitokelpoisuutta, allogeenisen kantasolujensiirron mahdollisuutta, ennustetta, ja selvitetään minimitaudin (MRD) seurantaan sopivat merkkiaineet (markkerit).
Anamneesi
- Infektio, vuoto-oireet, tukosoireet, CNS-tautiin viittaavat oireet
- Luustokivut
- Verenvuototaipumus, verisuonitukostaipumus (sic! riskitekijät)
- Tuumoreihin viittaavat oireet
- Säteilyaltistus, aiemmat solunsalpaajahoidot, liuotinaine- ja torjunta ainealtistus
- Sairaudet, jotka voivat vaikuttaa intensiivisen induktiohoidon sietoon (sydän, maksa, munuaiset, diabetes, neurologia, psyyke)
- Lääkitys ja allergiat
- < 66-vuotiaiden potilaiden sisarusten olemassaolo, terveydentila ja kotipaikka selvitetään mah-dollista allogeenista kantasolujensiirtoa ajatellen.
Kiininen tutkimus
- Infektiofokukset (suu, ruoansulatuskanava, perianaaliseutu, hengitystiet, virtsatiet, iho)
- Imusolmukestatus, maksan ja pernan status, iho, neurologinen status
- Verenvuodot, viitteet trombooseista
Veri- ja virtsanäytteet
- B-PVK+TKD, E-Retik, veriryhmä (E-ABORh), ristikoe (B-Xkoe), P-CRP, P-TT, P-APTT, P-D-dimeer, P-fibrinogeeni, P-AT3, P-FVIII, P-Alb, S-Prot, S-IgA, S-IgG, S-IgM, P-Krea, P-K, P-Na, P-Ca-ion, P-Pi, P-Mg, P‑Gluc, P-Bilir, P-Bilir‑kj, P-ASAT, P-ALAT, P-LD, P-AFOS, P-Uraat, S-CMVAb, S-EBVAb, S-ToxoAb, S-HIVAgAb, S-HAVAb, S-HBsAg, S-HCVAb, B-HbA1c, (f)P-kol ja (f)P-trigly, U-seula ja U –BaktV, raskaustesti (tarvittaessa)
- HLA‑kudostyypitysnäyte I (SPR: tutkimusnumero 5306, B-potilaan HLA1-tutkimuspaketti (kantasolujensiirto)) ja HLA‑kudostyypitysnäyte II (SPR: tutkimusnumero 5357 "B-potilaan HLA2-näyte pakastettavaksi myöhempää tutkimustarvetta varten"). TÄRKEÄÄ: nämä näytteet otetaan kahtena eri päivänä, mielellään diagnoosipäivänä ja sitä seuraavana päivänä. Lisätiedot: SPR Veripalvelu
- Veren kromosomitutkimus otetaan varalle ja tutkitaan, jos epäillään synnynnäistä poikkeavuutta luuytimen kromosomitutkimuksen perusteella tai muusta syystä
Luuydinnäytteet
Aspiraationäyte
- Sivelyvalmiste: MGG‑ ja rautavärjäys.
- Leukeemisten blastien immunofenotyypitys
- Luuytimen hematologinen kromosomitutkimus ja tarvittaessa moniväri-FISH-tutkimus
- Luuytimen hematologinen fuusiogeeniseulonta
Potilaat, joita ei voida hoitaa remissioon tähtäävällä hoidolla
- Luuytimen sivelyvalmiste ja immunofenotyypitys
- Luuytimen kromosomitutkimus
- Luuytimen hematologinen fuusiogeeniseulonta
Biopsianäyte
- Jos aspiraattinäytettä ei saada, otetaan luuydinbiopsia. Palanen biopsiasta kromosomitutkimukseen.
ALL:n geneettinen diagnoosi, portaittainen strategia:
1. Välittömästi dg-vaiheen näytteestä:
- Fuus-mR: BCR-ABL, ETV6-RUNX1, TCF3-PBX1, KMT2A(MLL)-AF4, SIL-TAL1 (pos. tulos varmistetaan FISH:llä)
- FISH: ABL1, ABL2, CSF1R/PDGRFB, MLL, TCF3-HLF(ja TCF3-PBX1), iAMP21(ETV6-RUNX1)
- Bm-Kromos (tai SNP/CGH-arraytutkimus suoraan, jollei kromosomitutkimusta tehdä)
- MLPA- (tai array-) tekniikalla seuraavat: BTG1, CDKN2A/B, EBF1, ETV6, IKZF1, PAX5, RB1, PAR1
- ASOAL-D (potilasspesifisten ASO-PCR-alukkeiden haku)
2. Toisen linjan tutkimukset alkuvaiheen tulosten perusteella:
- Jos Bm-Kromos epäonnistuu tai karyotyyppi on normaali, tehdään SNP/CGH-arraytutkimus ploidia-tason selvittämiseksi
- Jos BCR-ABL posit.: herkkä T315I-mutaatioanalyysi
- Jos FISH:n perusteella MLL- (KMT2A) uudelleenjärjestymä, tehdään jatkotutkimuksena uudelleenjärjestymän sekvensointi DNA-tasolta (lähetys Frankfurtiin)
- Jos ETP-ALL:
- FLT3-D
- myelooinen NGS-paneeli - Erillisen pyynnön perusteela, jos epäillään harvinaista fuusiota (esim. Ph-like, CRLF2, PAX5, KMT2A): RNAMut
Biopankkinäytteet (FHRB) ja rekisteröintisuostumus
- Pt-hembio (HUS-piiri) kaikilta potilailta, sisältää sekä luuydin- että verinäytteitä. Ks. tarkemmin: hematology.fi/fhrb.
- Kaikilta potilailta tulee pyytää lupa tietojen tallentamiseen (FHRB-tiedote ja suostumus)
- Muistettava näytteet myös remissio- ja relapsivaiheessa
Likvori
- Diagnoosivaiheessa ainoastaan keskushermosto- ja aivohermo-oireisilta potilailta. (Trombosyyttien oltava >40 x 109/l lumbaalipunktiota tehtäessä. Jos potilaan veressä on blasteja, lumbaalipunktion tekemistä on harkittava likvorin kontaminaatioriskin takia, vaikka riski onkin melko pieni).
Kuvantamistutkimukset
- Keuhkokuva, vatsan UÄ, harkiten vartalon TT
Muut näytteet
- EKG
Luokitus
- Kirjaudu tai rekisteröidy kommentoidaksesi
Luokitus
ICD-O
ICD-O
WHO:n luokitus ja vastaava ICD-O –koodi (SHR-muuttuja: icd-o-all)
|
B-lymfoblastinen leukemia/lymfooma, muuten määrittämätön |
9811/3 |
|
B-lymfoblastinen leukemia/lymfooma, johon liittyy t(9;22)(q34;q11.2); BCR-ABL1 |
9812/3 |
|
B-lymfoblastinen leukemia/lymfooma, johon liittyy t(v;11q23); MLL:n uudelleenjärjestymä |
9813/3 |
|
B-lymfoblastinen leukemia/lymfooma, johon liittyy t(12;21)(p13;q22); TEL-AML1 (ETV6-RUNX1) |
9814/3 |
|
B-lymfoblastinen leukemia/lymfooma, johon liittyy hyperdiploidia |
9815/3 |
|
B-lymfoblastinen leukemia/lymfooma, johon liittyy hypodiploidia (hypodiploidi-ALL) |
9816/3 |
|
B-lymfoblastinen leukemia/lymfooma, johon liittyy t(5;14)(q31;q32); IL3-IGH |
9817/3 |
|
B-lymfoblastinen leukemia/lymfooma, johon liittyy t(1;19)(q23;p13.3); E2A-PBX1 (TCF3-PBX1) |
9818/3 |
|
Burkittin soluleukemia |
9826/3 |
|
T-lymfoblastileukemia/lymfooma |
9837/3 |
Riskiluokitus
- Kirjaudu tai rekisteröidy kommentoidaksesi
Riskiluokitus
Suuri riski
Diagnoosivaiheessa
- Philadelphia-kromosomi/BCR-ABL1-positiivinen ALL (jos hoidon aikana jäännöstautipositiivinen, ks. hoito-ohje)
- t(4;11), MLL-AF4, t(17;19), TCF3-HLF
- Varhainen T-solulinjan ALL (blastit CD1a–negatiivisia)
- Kypsäsoluinen T-solulinjan ALL (blastit CD1a-negatiivisia)
Hoidon aikana
- 1. induktiolla ei päästä sytologiseen remissioon
- Jäännöstauti >0,01% (>1x10-4) 2. konsolidaatiohoidon jälkeen (virtaussytometria/PCR)
- Lisääntyvä jäännöstauti (1,0 log suurentuma toistetusti 2 vkon sisällä/ molekyläärinen relapsi)
Standardi riski
Muut kuin suuren riskin taudit.
Seuranta ja vastearvio
Seuranta ja vastearvio
Diagnoosivaiheessa tehtyjen tutkimusten perusteella valitaan potilaskohtaiset seurantatutkimukset (sytogenetiikka, molekyyligenetiikka, immunofenotyypitys), mikäli sellaisia on olemassa. Joka vastearviokerralla tulisi määrittää vähintään oheiset avainmuuttujat.
Seurannan avainmuuttujat (SHR)
| Kuvaus | Lyhenne | |
| 1 | Taudin status (tilanne-/vastearvio) | status-al |
| 2 | Blastien prosenttiosuus sivelyvalmisteesta | bm-blast-p |
| 3 | Blastien sytogeneettinen poikkeavuus (G-raita, FISH; %), jos olemassa | tutkimuskohtainen |
| 4 | Blastien spesifi immunofenotyyppi (%), jos olemassa | bm-mrd-facs-p |
| 5 | Blastien spesifi genotyyppi (%), jos olemassa | tutkimuskohtainen |
Seuranta-ajankohdat
1. Hypoplasiavaiheen arviointi induktiohoidon jälkeen, päivä +14
- Aspiraationäytteen morfologia tutkitaan hypoplasian asteen ja leukemian refraktorisuuden arvioimiseksi. Toisinaan remissio tulee kuitenkin hitaasti eikä päivän 14 näytteen perusteella voida päätellä lopullista refraktorisuutta. Jos blasteja on ylimäärä, tutkitaan luuydin myös päivinä 21 ja 28, jotta mahdollinen reinduktio ei turhaan viivästyisi. Jos ensimmäisellä induktiohoidolla ei päästä remissioon, hypoplasiavaiheen näyte tutkitaan myös toisen induktiohoidon jälkeen.
2. Vasteen arviointi induktiohoidon jälkeen, päivä +28
Luuydinnäyte tutkitaan verenkuvan korjaannuttua hoidon jälkeen (b-neut >1,0, b-trom >100) tai viimeistään päivänä 28. Tämän löydöksen sekä soluarvojen perusteella päätetään seuraavan hoidon aloittamisesta.
- Aspiraationäytteen morfologinen tutkimus, josta lasketaan blastien prosenttiosuus
- Diagnoosivaiheessa määritetty potilaskohtainen jäännöstaudin markkeri(t), mikäli sellainen on olemassa
Jos luuydin on päivänä 28 vielä hypoplastinen ja veressä on merkittävä sytopenia, eikä seuraavaa solunsalpaajahoitoa voida aloittaa, luuytimen toipumista ja remission saavuttamista seurataan aspiraationäytteen morfologian avulla noin viikon välein.
3. Vasteen arviointi konsolidaatiohoitojen aikana
Luuydinnäyte tutkitaan verenkuvan korjaannuttua jokaisen konsolidaatiohoidon jälkeen (b-neut >1,0, b-trom >100) tai viimeistään päivänä 28. Tämän löydöksen sekä soluarvojen perusteella päätetään seuraavan hoidon aloittamisesta.
- Aspiraationäytteen morfologinen tutkimus, josta lasketaan blastien prosenttiosuus
- Diagnoosivaiheessa määritetty potilaskohtainen jäännöstaudin markkeri(t), mikäli sellainen on olemassa
- Likvoritutkimukset tehdään hoito-ohjelman mukaisesti siten, että jokaisella lumbaalipunktiokerralla otetaan Li-solut ja Li-blastit -tutkimukset sekä erillisen harkinnan mukaan myös virtaussytometrinen solujen pintaproteiinianalyysi.
4. Vasteen arviointi konsolidaatiohoitojen päättymisen jälkeen
Granulosytopenian, trombosytopenian tai anemian kehittyminen tai paheneminen voivat viitata relapsiin ja ne samoin kuin blastien ilmaantuminen vereen, indisoivat luuytimen aspiraationäytteen tutkimisen. Erityisen tiiviisti on syytä seurata niitä potilaita, joiden relapsivaiheen hoidossa kantasolujensiirto voi tulla kyseeseen. Jos aiemmin jäännöstaudin suhteen negatiivisella potilaalla todetaan positiivinen tulos, ohjelmoidaan uusi näyte mahdollisimman pian. Merkittävän jäännöstaudin ilmaantuminen voi johtaa uusiin hoitoihin, allogeeniseen kantasolusiirtoon tai siirron jo saaneilla aikaistettuun immunosuppression purkuun ja lymfosyyttisiirtoon kantasolujen luovuttajalta (DLI).
Seurantaohjelma
1. vuosi konsolidaatiohoidon päättymisestä
- 1 kk välein B-PVK+TKD
- 3kk välein luuydinnäyte
- morfologia
- jäännöstauti (RQ-PCR ja tai FCM-menetelmällä)
2. vuosi konsolidaatiohoidon päättymisestä
- 3 kk välein B-PVK+TKD
- 3 kk välein luuytimen aspiraationäyte3kk välein luuydinnäyte
- morfologia
- jäännöstauti (RQ-PCR ja tai FCM-menetelmällä)
3.-4. vuosi (5 vuoteen saakka) konsolidaatiohoidon päättymisestä
- 4-6 kk välein PVK+TKD
- Luuytimen aspiraatiotutkimus tehdään, jos soluarvot tai oireet antavat siihen aihetta
Tutkimukset epäiltäessä relapsia
Granulosytopenian, trombosytopenian tai anemian kehittyminen tai paheneminen voivat viitata relapsiin ja ne samoin kuin blastien ilmaantuminen vereen, indisoivat luuytimen aspiraationäytteen tutkimisen. Erityisen tiiviisti on syytä seurata niitä potilaita, joiden relapsivaiheen hoidossa kantasolujensiirto voi tulla kyseeseen. ALL-potilaita seurattaessa on huomattava, että relapsi voi esiintyä myös pelkästään keskushermostossa.
Anamneesi:vuototaipumus, infektio, yleisoireet, kivut, päänsärky, pahoinvointi
Status:petekiat, mustelmat, infektiofokukset, hepatosplenomegalia, lymfadenopatia, tuumorit, iho, aivohermojen löydökset
Veri- ja virtsakokeet:B-PVK+TDK, CRP, P-Krea, P-K, P-Na, P-Bilir, P-ALAT, P-AFOS, P-TT, P‑Gluk, P-Alb, P-LD, P-Ca-ion, P-Pi, P-Mg, P-Uraat, B-Xkoe
Luuytimen aspiraationäyte, josta vähintään:
- MGG‑värjäys
- Kromosomitutkimus harkinnan mukaan
- Pintamerkkitutkimus harkinnan mukaan
- Jäännöstaudin seurantamarkkeri harkinnan mukaan
Relapsin määritelmä ja hoitoindikaatio:
- Molekyläärinen/immunofenotyyppien relapsi
- Morfologinen relapsi
Taudin status
- Kirjaudu tai rekisteröidy kommentoidaksesi
Taudin status
Vastearvion yhteydessä pyritään tautitaakan arvio ja taudin tilanne luokittelemaan oheisen taulukon mukaisesti (SHR-muuttuja: status-al). Taulukosta valitaan arviointihetkellä oleva paras vaste.
|
Koodi |
Lyhenne |
Nimike |
Määritelmä |
|
1 |
CR-MRDneg |
Remissio, jäännostauti negatiivinen |
|
|
2 |
CR-MRDpos |
Remissio, jäännostauti positiivinen |
|
|
3 |
CCR |
Sytogeneettinen remissio |
|
|
4 |
CR |
Morfologinen remissio |
|
|
5 |
CRi |
Remissio, puutteellinen luuytimen toipuminen |
|
|
6 |
PR |
Osittainen remissio |
|
|
7 |
Hypoplasia |
Dysplasia/hypoplasia |
|
|
8 |
RD |
Refraktaari tauti (RD) |
|
|
12 |
Relapsi (MRD) |
Molekyyligenettinen relapsi |
|
|
9 |
Relapsi (bm) |
Relapsi |
|
| 10 |
Relapsi (neuro) |
Isololoitu neurorelapsi |
|
| 11 |
Relapsi (ekstram) |
Isololoitu ekstramedullaarinen relapsi |
|
|
90 |
Exitus-relapsi |
Exitus, relapsi |
|
|
91 |
Exitus-infektio |
Exitus, infektio |
|
|
92 |
Exitus-aplasia |
Exitus, aplasia |
|
|
93 |
Exitus-muu tauti |
Exitus toisesta taudista johtuen |
|
| 98 | Exitus - ei hoitoa | Exitus, hoitoon ei mahdollisuutta | |
|
99 |
Exitus-tuntematon |
Exitus, syy tuntematon |
|
|
999 |
Poistunut seurannasta |
Seuranta loppunut (lost-to-follow-up) |
|
Hoito
Hoito
ALL-potilaat hoidetaan kunkin hoitoyksikön hoitolinjan mukaisesti
Helsingissä 16-45 –vuotiaat hoito-ohjelmaan soveltuvat potilaat saavat ensisijaisesti NOPHO-ALL2008-tutkimuksen mukaista hoitoa. Yli 45-vuotiaille tai NOPHO-ohjelmaan soveltumattomille käytetään ALL2000_amendement_2014 – hoito-ohjelmaa. BCR-ABL1/Ph–positiiviset ALL-taudit hoidetaan samoin periaattein siten, että heti kun tieto fuusiogeenistä on saatu, hoitoon liitetään dasatinibi 140 mg x1 2 viikon ajaksi, minkä jälkeen dasatinibin annos pienenee ad 100 mg x1 (ks. tarkempi ohje).
Ensilinjan hoito
Vaihtoehtoja:
- ALL2000_Amendment2014
- NOPHO ALL-2008 -tutkimushoito-ohjelma (TVH dos. Ulla Wartiovaara-Kautto/HYKS Syöpäkeskus)
- BCR-ABL1/Ph+ ALL-hoito-ohje
Refraktaarin taudin tai relapsivaiheen hoito
-
Jos tauti on refraktaari induktiohoidoille tai relapsoituu, pyritään tutkimukselliseen tai modifioituun hoitoon ja allogeeniseen kantasolusiirtoon
Mahdollisia hoitoja ovat mm.
- Tutkimushoito
- Klofarabiini-sytarabiini-etoposidi -hoito
- Nelarabiini (T-ALL)
- NOPHO ALL-2008 - blokkihoidot
- ALL2000_amendmentin konsolidaatiohoidot
| Liite | Koko |
|---|---|
| 36.22 KB |
Ph-positiivinen ALL
- Kirjaudu tai rekisteröidy kommentoidaksesi
Ph-positiivinen ALL
BCR-ABL1/Ph–positiiviset ALL-taudit hoidetaan joko ALL2000_amendment_2014 -hoito-ohjelman tai NOPHO-ALL2008 (induktiohoito non-HR) tutkimuksen protokollan (ei tutkimuspotilaana) mukaisesti siten, että heti kun tieto fuusiogeenistä on saatu, hoitoon liitetään tyrosiinikinaasiestäjä (TKE, dasatinibi 140 mg x1 2-4 viikon ajaksi, minkä jälkeen dasatinibin annos pienenee ad 100 mg x1, mikäli saavutettu vähintään hematologinen remissio).
TKE jatkuu vähintään ylläpitohoidon loppuun, jolloin se voidaan lopettaa, jos jäännöstautia ei ole todettavissa (MRD-negatiivinen). Allogeenisen siirron jälkeen TKE-lääkitys jatkuu 2 vuotta siirrosta. Dasatinibin tilalla voidaan myös käyttää imatinibia 600-800 mg/vrk; dasatinibin etuna on hyvä CNS-penetraatio, laajempi kinaasiestospektri ja siedettävyys, haittana pleuraeffuusioriski. Peruskorvattavuuden saamiseksi ensilinjan hoitona tulisi dasatinibia koskevaan B-lausuntoon lisätä maininta dasatinibin eduista imatinibi-lääkitykseen nähden (ks. templaattiteksti).
Ponatinibista on saatu viime vuosina hyviä alustavia hoitotuloksia yhdistettynä intensiiviseen solunsalpaajahoitoon (CVAD)(viite). Samoin blinatumomabi on tulossa myös Ph+ALL-taudin hoitoon (viite).
Allogeenista kantasolujensiirtoa harkitaan ensimmäisessä remissiossa potilailla, jotka täyttävät allogeenisen siirron soveltuvuuskriteerit. Tautitaakka tulisi olla mahdollisimman pieni ennen allogeenista siirtoa (MRD mielellään <0,01%).
HUS-hoito-ohje kevät 2017: Allogeenista kantasolujensiirtoa harkitaan potilailla, joilla hoidon jälkeen on merkittävä jäännöstauti (ALL2000_amend 1. kons.hoidon jälkeen MRD>0,01%; NOPHO-ALL2008 >5% d29, >0,01% d79; BCR-ABL1 RQ-PCR). Näiden aikapisteiden jälkeen allogeenista kantasolujensiirtoa harkitaan myös potilailla, joilla jäännöstauti jää tai muuttuu mitattavaksi (MRDpos).
Vanhemmilla tai huonokuntoisilla potilailla voidaan harkita myös dasatinibi-induktiohoitoa monoterapiana (tai deksametasonipulssihoitoon yhdistettynä), jolloin annos on 140 mg x1 kahden viikon ajan, minkä jälkeen 100 mg x1. Konsolidaatiohoitoon tulisi näilläkin potilailla dasatinibin oheen liittää solunsalpaajahoito dasatinibi-resistenttien kloonien ekspansion estämiseksi, esimerkiksi redusoituna ALL2000_amend2014-hoitoja ilman asparaginaasia ja it-hoitoja (jos TKE on dasatinibi).
Dasatinibin B-lausunto
- Kirjaudu tai rekisteröidy kommentoidaksesi
Dasatinibin B-lausunto
Dasatinibin peruskorvattavuutta koskevaan B-lausuntoon tulisi liittää maininta miksi imatinibi ei ole soveltuva lääke Ph+ALL-taudin hoidossa. Asiasta keskusteltu Kelan kanssa marraskuussa 2015. Esimerkki B-lausuntoon liitettävästä templaattitekstistä:
"Ph-positiivinen ALL on suuren riskin akuutti leukemia, jonka hoidossa on tärkeää tautitaakan nopea pienentäminen ja keskushermostotaudin tehokas esto ja hoito. Siksi imatinibi ei ole optimaalinen lääke, koska se ei läpäise veri-aivoestettä ja sen kinaasiestokirjo on kapea. Lisäksi suurella annoksella imatinibin siedettävyys on huonoa. Dasatinibi läpäisee veri-aivoesteen ja on osoitetusti tehokas neuroleukemian hoidossa. Sen kinaasiestokirjo on laaja, mikä on edullista korkean riskin taudin hoidossa. Lisäksi lääke on hyvin siedetty."
Kantasolujensiirto
Kantasolujensiirto
Allogeenista kantasolujensiirtoa harkitaan suuren relapsiriskin potilailla. Toisin kuin AML-taudissa, suurin osa potilaista voidaan hoitaa kemoterapialla ilman allogeenista kantasolujensiirtoa.
Tutkimuksia
- Kirjaudu tai rekisteröidy kommentoidaksesi
Tutkimuksia
ALL2000 (ensilinja)
ALL2000 (ensilinja)
Potilaan suostumus
Ennen hoidon aloittamista potilaalta pyydetään kirjallinen suostumus tutkimukseen osallistumisesta (liitetiedosto tämän sivun alalaidassa).
Yleistä
Hoidot 1–6 pyritään antamaan mahdollisimman nopeasti sen jälkeen, kun solut ovat toipuneet edellisestä hoidosta (B‑neutr ³1,0 x 109/l, B-tromb ³50 x 109/l). Solunsalpaajahoitoa aloitettaessa väliajan edelliseen G-CSF-hoitoon on oltava 72 t ja HD‑metotreksaattiin 7 pv. Jos kolmella hoidolla ei saavuteta remissiota, induktiohoito katsotaan epäonnistuneeksi ja jatkohoito on hoitavan lääkärin vapaasti valittavissa. Induktio- ja konsolidaatiohoitoja seuraa ylläpitohoito, joka jatkuu 3 vuoteen saakka diagnoosista.
Hoito 1
Suomen Leukemiaryhmän Akuutti leukemia -työryhmän kokouksessa 13.02.2013 päätettiin, ettei MEA vs. CVAD -satunnaistamista enää jatketa vaan kaikki potilaat saavat CVAD-induktiohoidon.
Hoito1, CVAD
Syklofosfamidi
300 mg/m2 x 2 30 minuutin infuusiona päivinä 1 - 3, yhteensä 6 annosta
Vinkristiini
1,4 mg/m2 (max 2 mg) nopeana infuusiona päivinä 4 ja 11, yhteensä 2 annosta
Doksorubisiini
50 mg/m2 30 min infuusiona päivänä 4.
Deksametasoni
20 mg/m2 po. päivinä 1 ‑ 4 ja 11 ‑ 14.
It. metotreksaatti
12,5 mg päivänä 8 (± 1 päivä), (trombosyyttiensiirto tarvittaessa niin, että B-tromb on yli 40 x 109/l). Huom! Tämä it.-hoito annetaan vain, kun CVAD on ensimmäisenä induktiohoitona.
It. sytarabiini
75 mg päivänä 22 (jos B-tromb yli 40 x 109/l).
It. metotreksaatti
12,5 mg päivänä 23 (jos B-tromb yli 40 x 109/l).
Huom!
Kun CVAD-hoito annetaan toisena hoitona (ks. alla), it. metotreksaattia ei anneta päivänä 8.
Päivien 22 ja 23 it.‑hoidot voidaan antaa aikaisemmin, jos trombosyytit ovat yli 40 x 109 tai ne annetaan myöhemmin, jos vaikea trombosytopenia vielä jatkuu yllämainittuina päivinä.
Jos virtsarakon seutuun on annettu sädehoitoa tai rakon limakalvo muutoin on vaurioitunut, syklofosfamidihoidon päivinä voidaan antaa mesnaa 600 mg/m2/vrk jatkuvana infuusiona.
Hoito 2
CVAD-hoito yllä esitetyn mukaisesti. It. metotreksaattia ei anneta päivänä 8.
Jos edellisestä hoidosta tuli virtsarakkovaivoja, syklofosfamidihoidon päivinä annetaan mesnaa 600 mg/m2/vrk jatkuvana infuusiona.
Hoito 3
Daunorubisiini
16‑55‑vuotiaat potilaat: 60 mg/m2 30 min infuusiona päivinä 1, 3 ja 5.
56‑65‑vuotiaat potilaat: 45 mg/m2 päivinä 1, 3, ja 5.
Vinkristiini
1,4 mg/m2 (max 2 mg) nopeana infuusiona päivinä 1 ja 8 sekä 0,4 mg/potilas/vrk jatkuvana infuusiona päivinä 15 ‑ 18 (4 vrk ajan).
Deksametasoni
20 mg/m2 po. päivinä 1 ‑ 4 ja 11 ‑ 14.
Asparaginaasi
Alle 56‑vuotaat potilaat 6000 yks/m2 ja 56 vuotta täyttäneet potilaat 4000 yks/m2 30 minuutin inf. päivinä 8, 11, 15, 18 ja 22.
It. sytarabiini
75 mg päivänä 22 (tai heti, kun B-tromb ovat yli 40 x 109/l).
Hoito 4
It. metotreksaatti
12,5 mg päivinä 1 ja 15.
Metotreksaatti
600 mg/m2 1 tunnin infuusiona ja 2400 mg/m2 23 tunnin infuusiona päivinä 1 ja 15 yhteensä 2 annosta.
Kalsiumfolinaatti
Kalsiumfolinaatti aloitetaan päivänä 2 ja päivänä 16, 30 tunnin kuluttua metotreksaatti‑infuusion alusta ja sitä annetaan po. tai iv. 5 annosta á 30 mg 4 tunnin välein. Sen jälkeen kalsiumfolinaattia annetaan po. 15 mg x 4 kahden vuorokauden ajan. Lisäksi kalsiumfolinaattia annetaan imeskeltäväksi ½ tablettia samoina ajankohtina. Virtsan alkalisoinnista on huolehdittava.
Ks Hematologinen kansio/Solunsalpaajiin liittyviä ohjeita/Metotreksaatti
Deksametasoni
20 mg/m2 po. päivinä 2 ‑ 5 ja päivinä 16 - 19
Vinkristiini
1,4 mg/m2 (max 2,0 mg/potilas) inf. päivinä 3 ja 17
Merkaptopuriini
60 mg/m2 po. päivinä 4 ‑ 13 (10 annosta) ja 18 - 25, yhteensä 8 annosta.
Jos päiväksi 15 suunniteltua metotreksaattihoitoa joudutaan siirtämään fulminantin infektion, vaikean mukosiitin tai vaikean neutropenian takia, kaikki päivien 15 ‑ 25 hoidot (metotreksaatti, kalsiumfolinaatti, vinkristiini, merkaptopuriini ja deksametasoni) siirtyvät blokkina yhtä monta päivää myöhäisemmiksi.
Hoito 5 pyritään aloittamaan päivänä 29.
Hoito 5
Doksorubisiini
30 mg/m2 30 minuutin infuusiona päivinä 1, 8 ja 15.
Vinkristiini
1,2 mg/m2 (max 2 mg/potilas) nopeana infuusiona päivinä 1, 8 ja 15.
Deksametasoni
10 mg/m2 po. päivinä 1 ‑ 14, minkä jälkeen deksametasoni lopetetaan viikon kuluessa.
It. sytarabiini
75 mg päivänä 1.
It. metotreksaatti
12,5 mg päivänä 2.
Syklofosfamidi
1000 mg/m2 30 minuutin inf. päivänä 29.
Tioguaniini
60 mg/m2 po. päivinä 29 - 42.
Sytarabiini
75 mg/m2 sc. päivinä 29-32 ja 36-39.
Hoito 6
Metotreksaatti
200 mg/m2 yhden tunnin infuusiona ja 800 mg/m2 23 tunnin infuusiona päivänä 1.
Kalsiumfolinaatti
Kalsiumfolinaattihoito aloitetaan 48 tunnin kuluttua metotreksaatti-infuusion alusta. Kerta-annoksena annetaan 50 mg po. ja sen jälkeen annetaan kuuden tunnin välein 15 mg po. kahdeksan annosta.
Ks Hematologinen kansio/Solunsalpaajiin liittyviä ohjeita/Metotreksaatti
Sytarabiini
Sytarabiinia annetaan kahden tunnin infuusiona 12 tunnin välein päivinä 2 ja 3, yhteensä 4 annosta.
16‑55‑vuotiaat potilaat: kukin kerta-annos on 3000 mg/m2 .
56‑65‑vuotiaat potilaat: kukin kerta-annos on 1000 mg/m2 .
Jos kreatiniinin tai alkalisen fosfataasin arvo on koholla, sytarabiinin annos on korkeintaan 1000 mg/m2.
Kortikosteroidisilmätipat
1 gtt kumpaankin silmään x 4 päivinä 2 ja 3.
CNS-profylaksi induktio- ja konsolidaatiohoitojen aikana
Intratekaalisesti annetaan sytarabiinia 75 mg ja metotreksaattia 12,5 mg hoito‑ohjelman mukaisina päivinä, edellyttäen että trombosyyttiarvo on yli 40 x 109/l. Lisäksi annetaan HD‑metotreksaatti- ja HD-sytarabiinihoitoa.
Sädehoito
Mediastinumin sädehoito
Mediastinumin sädehoito annetaan remissioon päästyä kaikille potilaille, joilla diagnoosin aikaan thorax‑kuvan perusteella on ollut osoitettavissa mediastinumin tuumori. Erityisesti T‑ALL:ssa mediastinumtuumori on tavallinen, mutta se voi esiintyä myös B‑solulinjan ALL:ssa.
Muiden tuumorialueiden sädehoito
Tuumorialueen sädehoito annetaan remissioon päästyä kaikille potilaille, joilla diagnoosin aikaan on todettu yli 5 cm läpimittainen tuumori.
Testisten sädehoito
Testisten sädehoito 24 Gy annetaan niille potilaille, joille testisleukemiaepäilyn takia on tehty biopsia ja todettu testiksen leukeeminen infiltraatio.
Kantasolujensiirtopotilaat
Jos allogeeninen kantasolujensiirto voi tulla kyseeseen, on syytä neuvotella siirtokeskuksen kanssa ennen sädehoidon antamista.
Ylläpitohoito
Ylläpitohoito aloitetaan heti solujen toivuttua hoidosta 6 ja sitä jatketaan kunnes diagnoosipäivästä on kulunut 3 vuotta tai ilmaantuu relapsi. Ylläpitohoidon annoksia lisätään tai vähennetään siten, että veren leukosyyttien määrä pysyttelee 3,0 x 109/l tienoilla ja neutrofiilien määrä yli 1,0 x 109/l. Relapsien välttämiseksi on tärkeää pyrkiä tehokkaaseen ylläpitohoitoon.
Merkaptopuriini
60 mg/m2 x 1 po. päivittäin.
Metotreksaatti
20 mg/m2 po. yhtenä annoksena kerran viikossa.
Vinkristiini
1,4 mg/m2 (max 2 mg/potilas) nopeana infuusiona neljän viikon välein edellisen hoidon alusta seuraavan hoidon alkuun laskettuna. Vinkristiinihoito jatkuu 12 kk:n ajan. Jos potilaalla on merkittävä neuropatia, vinkristiini jätetään antamatta.
Prednisoni
60 mg/m2 po. viiden päivän ajan neljän viikon välein edellisen hoidon alusta seuraavan hoidon alkuun laskettuna. Hoito jatkuu 12 kk:n ajan.
CNS‑profylaksi ylläpitohoidon aikana
CNS‑profylaksina annetaan metotreksaattia 12,5 mg it. suuren riskin potilaille 6 kertaa ja muille potilaille 3 kertaa.
Suuren riskin potilaat
Jos potilaalla on diagnoosivaiheessa
1) LD yli 1175 U/l (5 x normaalin yläraja)
2) B-leuk yli 50 x 109/l tai
3) ekstramedullaarisia infiltraatioita muualla kuin pernassa, maksassa tai imusolmukkeissa
it. hoitoja annetaan yhteensä kuusi kertaa 4 viikon välein.
Potilaat, joilla ei ole suuren riskin tekijöitä diagnoosivaiheessa
Jos potilaalla ei ole edellä mainittuja neuroleukemian riskitekijöitä, it.-hoitoja annetaan yhteensä kolme kertaa 8 viikon välein
AML
AML
 Akuutti myelooinen leukemia (AML) ryhmä geneettisesti erilaisia veritauteja, joiden yhteisenä piirteenä on kypsymishäiriö ja hallitsematon kasvu luuytimen myelooisissa progenitori- ja kantasoluissa. AML:n diagnoosi perustuu luuytimen (tai veren) morfologiaan ja leukemiaspesifisien pintaproteiinien ja sytomolekylaaristen poikkeavuuksien osoittamiseen.
Akuutti myelooinen leukemia (AML) ryhmä geneettisesti erilaisia veritauteja, joiden yhteisenä piirteenä on kypsymishäiriö ja hallitsematon kasvu luuytimen myelooisissa progenitori- ja kantasoluissa. AML:n diagnoosi perustuu luuytimen (tai veren) morfologiaan ja leukemiaspesifisien pintaproteiinien ja sytomolekylaaristen poikkeavuuksien osoittamiseen.
AML-tautia hoidetaan monisolunsalpaajahoidolla ja osalla potilaista hoitoa tehostetaan allogeenisen kantasolujensiirron avulla. Hoito perustuu Suomen Leukemiaryhmän AML-2012 hoito-ohjelmaan niillä potilailla, joille voidaan antaa intensiivistä, remissioon tähtäävää solunsalpaajahoitoa. Muilla potilailla hoidon aiheet ja laatu punnitaan potilaskohtaisesti.
Sivuto on siirtymässä toiseen osoitteeseen. Voimassa olevat ohjeet löytyvät: https://www.hematology.fi/fi/hoito-ohjeet/veritaudit/akuutit-leukemiat/a...
| Toimitus | Pirjo Koistinen, Kimmo Porkka, Erkki Elonen, Mika Kontro, Riikka Räty |
Diagnoosi
Diagnoosi
Sivusto siirtymässä toiseen osoitteseen. Ajankohtaiset ohjeet löytyvät toistaiseksi linkistä: https://www.hematology.fi/fi/hoito-ohjeet/veritaudit/akuutit-leukemiat/a...
AML-taudin diagnoosi perustuu luuytimen (tai veren) morfologiseen tutkimukseen, sekä blastien virtaussytometriseen pintaproteiinitutkimukseen, joita täydennetään myöhemmin valmistuvilla sytomolekyyligeneettisillä tutkimuksilla.
Diagnostiset kriteerit (WHO2008): blastien määrä veressä ja/tai luuytimessä >=20% (tarkennukset).
Diagnoosivaiheen tutkimusten tulosten perusteella arvioidaan potilaan hoitokelpoisuutta, allogeenisen kantasolujensiirron mahdollisuutta, ennustetta ja selvitetään minimitaudin (MRD) seurantaan sopivat merkkiaineet (markkerit).
Anamneesi
- infektio‑ ja vuoto-oireet
- luustokivut
- tuumoreihin viittaavat oireet
- säteilyaltistus, aiemmat solunsalpaajahoidot, liuotinaine- ja torjunta‑ainealtistus
- sairaudet, jotka voivat vaikuttaa intensiivisen induktiohoidon sietoon (sydän, maksa, munuaiset, diabetes, neurologia, psyyke)
- lääkitys ja allergiat
- alle 66‑vuotiaiden potilaiden sisarusten olemassaolo, terveydentila ja kotipaikka selvitetään mahdollista allogeenista kantasolujensiirtoa ajatellen.
Kliininen tutkimus
- infektiofokukset (suu, ruoansulatuskanava, perianaaliseutu, hengitystiet, virtsatiet, iho)
- verenvuototaipumus
- imusolmukestatus, maksan ja pernan status, ihoinfiltraatiot ja ienhyperplasia.
Veri- ja virtsanäytteet
- B-PVK+TKD, E-Retik, veriryhmä (E-ABORh), ristikoe (B-Xkoe), P-CRP, P-TT, P-APTT, P-D-dimeer, P-fibrinogeeni, P-Alb, S-Prot, S-IgA, S-IgG, S-IgM, P-Krea, P-K, P-Na, P-Ca-ion, P-Pi, P-Mg, P‑Gluc, P-Bilir, P-Bilir‑kj, P-ASAT, P-ALAT, P-LD, P-AFOS, P-Uraat, S-CMVAb, S-EBVAb, S-ToxoAb, S-HIVAgAb, S-HAVAb, S-HBsAg, S-HCVAb, B-HbA1c, (f)P-kol ja (f)P-trigly, U-seula ja U –BaktV, raskaustesti (tarvittaessa).
- 8 ml verta CPT-putkeen molekyyligeneettisiä tutkimuksia varten (TyksLab)
- HLA‑kudostyypitysnäyte I (SPR: tutkimusnumero 5306, B-potilaan HLA1-tutkimuspaketti (kantasolujensiirto)) ja HLA‑kudostyypitysnäyte II (SPR: tutkimusnumero 5357 "B-potilaan HLA2-näyte pakastettavaksi myöhempää tutkimustarvetta varten"). TÄRKEÄÄ: nämä näytteet otetaan kahtena eri päivänä, mielellään diagnoosipäivänä ja sitä seuraavana päivänä. Lisätiedot: SPR Veripalvelu
Luuydinnäytteet
Aspiraationäyte
- Sivelyvalmiste: MGG‑, ANAE‑ ja rautavärjäykset.
- Leukeemisten blastien immunofenotyypitys
- Luuytimen hematologinen kromosomitutkimus ja tarvittaessa moniväri-FISH-tutkimus
- Luuytimen hematologinen fuusiogeeniseulonta
- FISH-seulonta: EVI, MLL ja kromosomien 5 ja 7 deleetiot (5q31, 7q31, kr. 7 sentromeeri)(vain TaYS ja TYKS)
- FLT3-geenin mutaatiotutkimus ja nukleofosmiinigeenin (NPM1) mutaatiotutkimus
- CEBPA-geenin mutaatiotutkimus; ainoastaan potilailla, joilla ei muita markkereita (normaali sytogenetiikka, ei FLT3- tai NPM1-mutaatioita)
- NUP98-NSD1-fuusiogeenitutkimus (fuusiogeeniseula II, TYKS)
- TyksLabiin lähetettävät molekyyligeneettiset näytteet otetaan kahteen 4ml:n CPT-putkeen
- Molekyyligeneettisten tutkimusten porrastus (TYKS)
70 vuotta täyttäneet sekä potilaat joita ei voida hoitaa remissioon tähtäävällä hoidolla
- luuytimen sivelyvalmiste ja immunofenotyypitys
- luuytimen kromosomitutkimus
Biopsianäyte
-
Jos aspiraattinäytettä ei saada niin otetaan luuydinbiopsia. Palanen biopsiasta kromosomitutkimukseen.
AML:n geneettinen diagnoosi, portaittainen strategia:
1. Välittömästi dg-vaiheen näytteestä:
- FISH: 5q-, 7q-, EVI1, TP53del, MLL
- Bm-Kromos: (tarv. varmistetaan FISH:llä)
- Fuus-mR: (pos. löydös varmistus FISHillä) PML-RARA, CBFB-MYH11, RUNX1-RUNX1T1, BCR-ABL
- FLT3-D
- Myeloinen NGS-paneeli (sis. NPM1, KIT ym.)
2. Mikäli Fuu-mR negatiivinen:
- Fuus2-mR:MLL-MLLT3, DEK-NUP214, NUP98-NSD1
3. Erillisestä pyynnöstä, kun sytogenetiikan perusteella epäily fuusiogeenistä tai mikäli Fuus-mR, NPM1 ja Fuus2-mR kaikki negatiivisia:
- RNAMut
- Erillisen pyynnön perusteella, mikäli ei huonon ennusteen löydöksiä: CEBPA-geenin
mutaatioanalyysi
Biopankkinäytteet (FHRB) ja rekisteröintisuostumus
- Pt-hembio (HUS-piiri) kaikilta potilailta, sisältää sekä luuydin- että verinäytteitä. Ks. tarkemmin: hematology.fi/fhrb
- Kaikilta AML-2012-hoito-ohjelmaan osallistuvilta (miel. myös muilta) potilailta tulee pyytää lupa tietojen tallentamiseen (FHRB-tiedote ja suostumus)
Likvori
- Ainoastaan keskushermosto‑ ja aivohermo‑oireisilta potilailta
- Suuren riskin potilailta remissioon päästyä
Trombosyyttien on oltava >40 x 109/l lumbaalipunktiota tehtäessä. Jos potilaan veressä on blasteja, lumbaalipunktion tekemistä on harkittava likvorin kontaminaatioriskin takia, vaika riski onkin melko pieni.
Kuvantamistutkimukset
- Keuhkokuva, vatsan UÄ, harkiten vartalon TT-kuvaus
Muut näytteet
- EKG
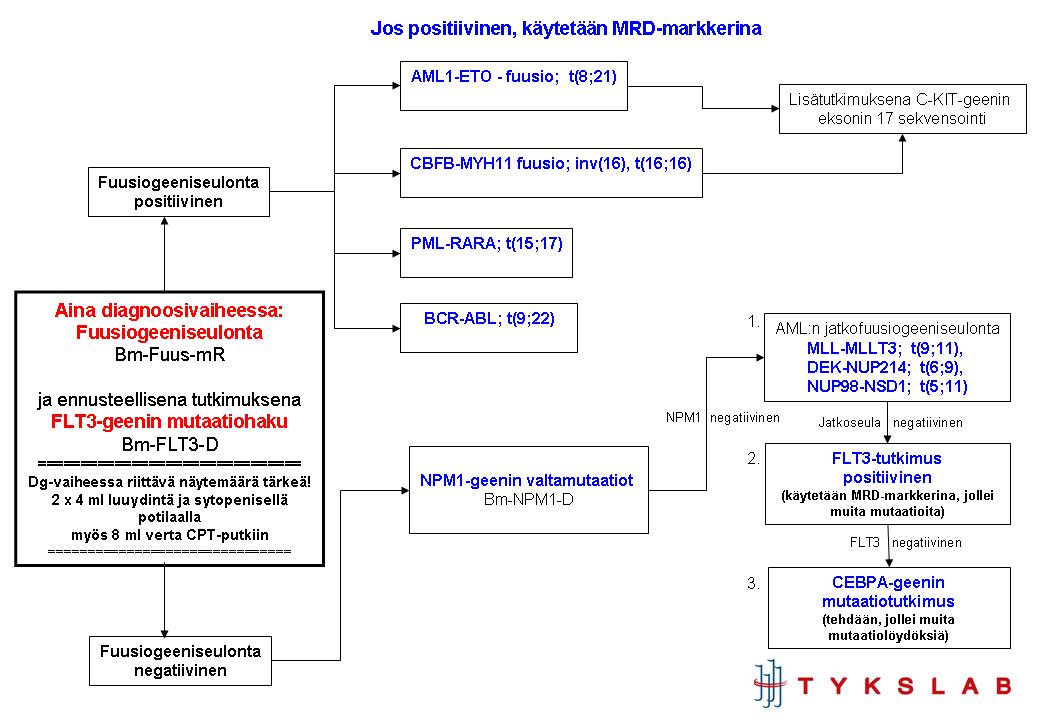
Molekyyligeneettisten tutkimusten porrastus (TYKSLAB), päivitys kesken
- Kirjaudu tai rekisteröidy kommentoidaksesi
Molekyyligeneettisten tutkimusten porrastus (TYKSLAB), päivitys kesken
Luokitus
- Kirjaudu tai rekisteröidy kommentoidaksesi
Luokitus
Sivusto siirtymässä toiseen osoitoitteseen. Ajankohtaiset ohjeet löytyvät toistaiseksi linkistä: https://www.hematology.fi/fi/hoito-ohjeet/veritaudit/akuutit-leukemiat/a...
ELN 2017 riskiluokitus
ELN 2017 riskiluokitus
AML-2018 -protokollan geneettinen riskiluokitus on seuraavanlainen (perustuu European LeukemiaNetin suositukseen v. 2017):
Pieni riski (ryhmä I)
Seuraavat karyotyyppipoikkeavuudet (yksin tai muiden poikkeavuuksien yhteydessä)
- t(8;21) (q22;q22.1) [ RUNX1-RUNX1T1]
- inv16 (p13.1q22)/t(16;16) (p13.1;q22) [CBFB-MYH11]
- NPM1-mutaatio ilman Flt3-ITD-mutaatiota tai kun Flt3-ITD-alleelikuorma on matala (<0,5)
- kaksoismutatoitunut CEBPA ilman Flt3-ITD-mutaatiota
Keskiriski (Ryhmä II)
Ei korkean riskin karyotyyppimuutoksia tai mutaatioita seuraavien mutaatioiden yhteydessä:
- Korkean alleelikuorman (>0,5) FLT3-ITD-mutaatio ja NPM1-mutaatio
- Matalan alleelikuorman (<0,5) FLT3-ITD-mutaatio ilman NPM1-mutaatiota
Normaali karyotyyppi ilman FLT3-ITD- ja NPM1-mutaatioita
Seuraavat ryhmiin I ja III kuulumattomat karyotyyppipoikkeavuudet
- t(9;11)(p21.3;q23.3) [MLLT3-KMT2A]
Muut ryhmään I ja III kuulumattomat karyotyyppipoikkeavuudet
Suuri riski (Ryhmä III)
Seuraavat karyotyyppipoikkeavuudet
- t(6;9)(p23;q34.1) [DEK-NUP214]
- t(v;11q23.3) [uudelleenjärjestäytynyt KMT2A luk.ottamatta muutosta t(9;11)]
- t(9;22)(q34.1;q11.2) [BCR-ABL1]
- inv(3)(q21.3q26.2) / t(3;3)(q21.3;q26.2) [GATA2, MECOM(EVI1)]
- -5, 5q-, -7, -17/poikkeava 17p
- kompleksinen karyotyyppi (kolme tai useampi karyotyyppipoikkeavuus. Lukuun ei lasketa: t(15;17), t(8;21), inv(16), t(16;16), t(9;11), t(v;11)(v;q23.3), t(6;9), inv(3), t(3;3), BCR-ABL1)
- monosomaalinen karyotyyppi (yksi autosomin monosomia + yksi tai useampi rakenteellinen karyotyyppipoikkeavuus; kaksi tai useampi autosomin monosomiapoikkeavuutta)
Huom! Pienen riskin karyotyyppipoikkeavuuksia ja XY monosomioita ei lasketa mukaan
Seuraavat korkean riskin mutaatiot
- korkean alleelikuorman (>0,5) Flt3-ITD -mutaatio ilman NPM1-mutaatiota
- RUNX1 -mutaatio (ei pienen riskin muutoksiin yhdistyneenä)
- ASXL1 -mutaatio (ei pienen riskin muutoksiin yhdistyneenä)
- TP53 -mutaatio (esiintyy yleensä kompleksisen tai monosomaalisen karyotyypin kanssa)
ICD-O
ICD-O
WHO:n alaluokitus ja vastaava ICD-O -koodi (SHR-muuttuja: icd-o-aml)
| AML, johon liittyy spesifinen toistuva geneettinen poikkeavuus | ICD-O | |
| AML ja t(8;21)(q22;q22); RUNX1-RUNX1T1 | 9896/3 | |
| AML ja inv(16)(p13.1q22) tai t(16;16)(p13.1;q22); CBFB-MYH11 | 9871/3 | |
| APL ja t(15;17)(q22;q12); PML-RARA* (FAB M3) | 9866/3 | |
| AML ja t(9;11)(p22;q23); MLLT3-MLL** | 9897/3 | |
| AML ja t(6;9)(p23;q34); DEK-NUP214 | 9865/3 | |
| AML ja inv(3)(q21q26.2) tai t(3;3)(q21;q26.2); RPN1-EVI1 | 9869/3 | |
| AML (megakaryoblastinen) ja t(1;22)(p13;q13); RBM15-MKL1 | 9911/3 | |
| AML ja mutatoitunut NPM1 (ehdollinen) | 9861/3 | |
| AML ja mutatoitunut CEBPA (ehdollinen) | 9861/3 | |
| AML, johon liittyy myelodysplastisia muutoksia*** | 9895/3 | |
| Aikaisempiin hoitoihin liittyvä AML # | 9920/3 | |
| Muutoin spesifioimaton AML | 9861/3 | |
| AML ja minimaalinen erilaistuminen (FAB M0) | 9872/3 | |
| AML ilman kypsymistä (FAB M1) | 9873/3 | |
| AML ja vähäinen kypsyminen (FAB M2) | 9874/3 | |
| AML, myelomonosyyttileukemia (FAB M4) | 9867/3 | |
| AML, monoblastinen/monosyyttinen leukemia (FAB M5a/M5b) | 9891/3 | |
| AML, akuutti erytroleukemia (FAB M6) | 9840/3 | |
| Puhdas erytroleukemia, Erytroleukemia (erytroinen/myelooinen) | ||
| AML, akuutti mekakaryoblastileukemia (FAB M7) | 9910/3 | |
| AML, akuutti basofiilinen leukemia | 9870/3 | |
| AML, panmyeloosi ja myelofibroosi (= akuutti myelofibroosi/myeloskleroosi) | 9931/3 | |
| Ekstramedullaarinen AML = myelooinen sarkooma, klorooma, | 9930/3 | |
| granulosyyttinen sarkooma) | ||
| Downin syndroomaan liittyvä AML | 9898/3 | |
| Blastinen plasmasytoidinen dendriittisoluleukemia | 9727/3 | |
| Akuutti leukemia, erilaistumislinja kaksisuuntainen/epäselvä | ||
| Akuutti erilaistumaton leukemia | 9801/3 | |
| Akuutti leukemia, sekafenotyyppi ja t(9;22)(q34;q11.2); BCR-ABL1 ## | 9806/3 | |
| Akuutti leukemia, sekafenotyyppi ja t(v;11q23); MLL:n uudelleenjärjestymä | 9807/3 | |
| Akuutti leukemia, sekafenotyyppi, B/myelooinen, muutoin spesifioimaton | 9808/3 | |
| Akuutti leukemia, sekafenotyyppi, T/myelooinen, muutoin spesifioimaton | 9809/3 | |
| NK solu lymfoblastinen leukemia/lymfooma (ehdollinen) | ||
*Muut RARA:aan liittyvät translokaatiot ilmaistaan erikseen: t(11;17)(q23;q12)=ZBTB16-RARA,
t(11;17)(q13;q12)=NUMA1-RARA, t(5;17)(q35;q12)=NPM1-RARA, tai STAT5B-RARA
**Muut MLL:aan liittyvät translokaatiot ilmaistaan erikseen: t(6;11)(q27;q23)=MLLT4-MLL, t(11;19)(q23;p13.3)=MLL-MLLT1, t(11;19)(q23;p13.1) =MLL-ELL, t(10;11)(p12;q23) =MLLT10-MLL
***>20 % blasteja veressä tai luuytimessä ja mikä tahansa seuraavista: aikaisempi MDS tai MDS/MPD, yli 50 % soluista vähintään kahdessa solulinjassa on dysplastisia, tai tautiin liittyy myelodysplasialle ominainen sytogeneettinen poikkeavuus, joita ovat: kompleksinen karyotyyppi (kolme tai useampi kromosomaalinen poikkeavuus), balansoimattomat löydökset: -7, del(7q), -5, del(5q), i(17q), i(17p), -13, del(13q), del(12p), t(12p), del(9q), idic(X)(q13), balansoidut löydökset: t(11;16)(q23;p13.3), t(3;21)(q26.2;q22.1), t(1;3)(p36.3;q21.1), t(2;11)(p21;q23), t(5;12)(q33;p12), t(5;7)(q33;q11.2), t(5;17)(q33;p13), t(5;10)(q33;q21), t(3;5)(q25;q34).
# Altistuminen sytotoksisille aineille: alkyloivat aineet, ionisoiva sädehoito, topoisomeraasi II:n inhibiittorit, muut.
## BCR-ABL1 positiivinen leukemia voi olla sekasolulinjainen akuutti leukemia, mutta se tulisi hoitaa kuten BCR-ABL1 positiivinen ALL.
ELN 2010 riskiluokitus
ELN 2010 riskiluokitus
AML-2012-tutkimuksen geneettinen riskiluokitus on seuraavanlainen (perustuu European LeukemiaNet:in suositukseen v. 2010):
Pieni riski (ryhmä I)
Seuraavat karyotyyppipoikkeavuudet (yksin tai muiden poikkeavuuksien yhteydessä)
- t(8;21) (q22;q22) [ RUNX1-RUNX1T1]
- inv16 (p13.1;q22)/t(16;16) (p13.1;q22) [CBFB-MYH11]
Normaali karyotyyppi seuraavien mutaatioiden yhteydessä
- NPM1-mutaatio ilman Flt3-ITD-mutaatiota
- kaksoismutatoitunut CEBPA ilman Flt3-mutaatiota
Keskiriski (Ryhmä II)
Normaali karyotyyppi seuraavien mutaatioiden yhteydessä:
- FLT3-ITD-mutaatio ja NPM1-mutaatio
- FLT3-ITD-mutaatio ilman NPM1-mutaatiota
Normaali karyotyyppi ilman FLT3-ITD- ja NPM1-mutaatioita
Seuraavat ryhmään I ja III kuulumattomat karyotyyppipoikkeavuudet
- t(9;11)(p22;q23); MLLT3-MLL
- muut ryhmään I ja III kuulumattomat karyotyyppipoikkeavuudet
Suuri riski (Ryhmä III)
Seuraavat karyotyyppipoikkeavuudet
- inv(3)(q21q26.2) / t(3;3)(q21;q26.2) [RPN1-EVI1]
- t(6;9)(p23;q34) [DEK-NUP214]
- t(v;11(v;q23) [uudelleenjärjestäytynyt MLL luk.ottamatta muutosta t(9;11)]
- -5, 5q-, -7, poikkeava 17p
- kompleksinen karyotyyppi (kolme tai useampi karyotyyppipoikkeavuus. Lukuun ei lasketa seuraavia: t(15;17), t(8;21), inv(16), t(16;16), t(9;11), t(v;11)(v;g23), t(6;9), inv(3), t(3;3))
- monosomaalinen karyotyyppi (yksi autosomin monosomia + yksi tai useampi rakenteellinen karyotyyppipoikkeavuus; kaksi tai useampi autosomin monosomiapoikkeavuutta)
Huom! Pienen riskin karyotyyppipoikkeavuuksia ja XY monosomioita ei lasketa mukaan
Seuranta ja vastearvio
Seuranta ja vastearvio
Sivusto siirtymässä toiseen osoitoitteseen. Ajankohtaiset ohjeet löytyvät toistaiseksi linkistä: https://www.hematology.fi/fi/hoito-ohjeet/veritaudit/akuutit-leukemiat/a...
Avainmuuttujat
Hoitotulosten arviointia varten potilaiden diagnoosivaiheen tietoja, annetut hoidot ja niiden vasteet kerätään Suomen hematologiseen rekisteriin (SHR) potilaan suostumuksella. AML-2012 ohjelmaan (vähimmäisvaatimuksena) kirjattavat tiedot on merkitty peukalomerkillä ( ).
Induktio‑ ja konsolidaatiovaiheen tutkimukset
Veritutkimukset hypoplasiavaiheessa
- Vähintään kolmesti viikossa: B-PVK+Ne ja P-CRP.
- Vähintään kahdesti viikossa: B-XKoe, P-Krea, P-K, P-Na, P-Bilir, P-ALAT, P-AFOS.
- Vähintään kerran viikossa: P-TT/INR, P‑Gluk, P-Alb, P-LD, P-Ca-ion, P-Pi, P-Mg, P-Uraat (vain ensimmäisen hoidon 2 ensimmäisen viikon aikana)
- Solujen toipumisvaiheessa on tärkeää tutkia blastien esiintymistä veressä: B-PVK+TKD
- Kuumeisten, pahoinvoivien ja ripuloivien potilaiden veren soluarvoja, CRP:tä, elektrolyyttejä, happo-emästasetta ja kreatiniinia seurataan päivittäin
- Infektionäytteiden aktiiviseen ottamiseen ja oikea‑aikaiseen lähettämiseen on kiinnitettävä erityistä huomiota (neutropeenisen potilaan infektiot)
Luuydinnäytteet induktiohoidon jälkeen
Hypoplasiavaihe induktiohoidon jälkeen, päivä 14 (suositus, ei kuulu AML2012-ohjelmaan)
- Aspiraationäytteen morfologinen näyte tutkitaan hypoplasian asteen ja leukemian refraktorisuuden arvioimiseksi. Toisinaan remissio tulee kuitenkin hitaasti eikä päivän 14 näytteen perusteella voida päätellä lopullista refraktorisuutta. Jos blasteja on ylimäärä, tutkitaan luuydin myös päivinä 21 ja 28, jotta mahdollinen reinduktio ei turhaan viivästyisi.
- Jos ensimmäisellä induktiohoidolla ei päästä remissioon, hypoplasiavaiheen näyte tutkitaan myös toisen induktiohoidon jälkeen.
Remission arviointi solujen toipuessa induktiohoidon jälkeen, päivä 28
Luuydinnäyte tutkitaan remission arvioimiseksi tutkitaan verisolujen noustessa hoidon jälkeen tai viimeistään päivänä 28. Tämän löydöksen sekä soluarvojen perusteella päätetään seuraavan hoidon aloittamisesta.
- Jos luuydin on vielä hypoplastinen ja veressä on merkittävä sytopenia, eikä seuraavaa solunsalpaajahoitoa voida aloittaa, luuytimen toipumista ja remission saavuttamista seurataan aspiraationäytteen morfologian avulla noin viikon välein.
- Tässä vaiheessa otetaan näyte (näytteet) myös jäännöstaudin arvioimiseksi, jos markkeri on.
Likvoritutkimus remission varmistamiseksi induktiohoidon jälkeen seuraavissa tilanteissa:
- DG-vaiheessa sytogeneettisenä muutoksena kromosomi 16:n inversio tai t(16;16)
- Dg-vaiheen B-leuk > 100 x 109/l
- Dg-vaiheessa ekstramedullaarinen tauti muualla kuin keskushermostossa
Luuydinnäytteet konsolidaatiohoidon jälkeen
Jokaisen konsolidaatiohoidon jälkeen otetaan luuydinnäytteet morfologisen remission ja jäännöstaudin arvioimiseksi.
Seurantavaiheen tutkimukset (AML-2012)
Luuydinnäytteestä tutkitaan allogeeniseen kantasolusiirtohoitoon soveltuvilta pienen riskin potilailta morfologian lisäksi jäännöstauti (RQ-PCR / FC-menetelmällä) 2 vuoteen saakka. Jos aiemmin jäännöstaudin suhteen negatiivisella potilaalla todetaan positiivinen tulos, ohjelmoidaan uusi näyte mahdollisimman pian.
- Merkittävän jäännöstaudin ilmaantuminen voi johtaa allogeeniseen kantasolusiirtoon tai siirron jo saaneilla aikaistettuun immunosuppression purkuun ja lyfosyyttisiirtoon kantasolujen luovuttajalta (DLI)
- Muissa riskiryhmissä jäännöstautiseuranta hoitavan yksikön harkinnan mukaisesti
- Uusi solunsalpaajahoito aloitetaan vasta kun todetaan morfologinen relapsi
Seurantaohjelma
1. vuosi konsolidaatiohoidon päättymisestä
- 1 kk välein B-PVK+TKD
- 3 kk välein luuytimen aspiraationäyte
- morfologia
- jäännöstauti (RQ-PCR ja tai FC-menetelmällä): allogeenisen kantasolusiirron piirissä olevilta pienen riskin potilailta, myös muissa riskiryhmissä suositeltava
2. vuosi konsolidaatiohoidon päättymisestä
- 3 kk välein B-PVK+TKD
- 3 kk välein luuytimen aspiraationäyte
-
- morfologia
- jäännöstauti (RQ-PCR ja tai FC-menetelmällä): allogeenisen kantasolusiirron piirissä olevilta pienen riskin potilailta, myös muissa riskiryhmissä suositeltava
-
3.-4. vuosi (5 vuoteen saakka) konsolidaatiohoidon päättymisestä
- 4 - 6 kk välein B-PVK+TKD
- Luuytimen aspiraatiotutkimus tehdään, jos soluarvot tai oireet antavat siihen aihetta
Seurantapaikka
- HUS: Remissiossa olevat HUS:n piirin potilaat seurataan 5 v ajan hoitojen päättymisestä HUS:n sairaaloissa ja sen jälkeen, jos erityisiä ongelmia ei ole, TK:ssa tai muun oman lääkärin toimesta. Muiden keskusairaaloiden alueiden potilaat seurataan ao. keskussairaalan toimesta, kun konsolidaatiohoidot on annettu.
Tutkimukset epäiltäessä relapsia
Granulosytopenian, trombosytopenian tai anemian kehittyminen tai paheneminen voivat viitata relapsiin ja ne samoin kuin blastien ilmaantuminen vereen, indisoivat luuytimen aspiraationäytteen tutkimisen. Erityisen tiiviisti on syytä seurata niitä potilaita, joiden relapsivaiheen hoidossa kantasolujensiirto voi tulla kyseeseen.
Anamneesi: vuototaipumus, infektio, yleisoireet, kivut, päänsärky, pahoinvointi
Status: petekiat, mustelmat, infektiofokukset, hepatosplenomegalia, lymfadenopatia, tuumorit, ihoinfiltraatio, ienhyperplasia, aivohermojen löydökset
Veri- ja virtsakokeet: B-PVK+TDK, CRP,P-Krea, P-K, P-Na, P-Bilir, P-ALAT, P-AFOS, P-TT, P‑Gluk, P-Alb, P-LD, P-Ca-ion, P-Pi, P-Mg, P-Uraat, B-Xkoe
Luuytimen aspiraationäyte:
- MGG‑värjäys, muut värjäykset harkinnan mukaan
- Kromosomitutkimus harkinnan mukaan
- Pintamerkkitutkimus harkinnan mukaan
- Jäännöstaudin seurantamarkkeri harkinnan mukaan
Taudin status
- Kirjaudu tai rekisteröidy kommentoidaksesi
Taudin status
Vastearvion yhteydessä pyritään tautitaakan arvio ja taudin tilanne luokittelemaan oheisen taulukon mukaisesti (SHR-muuttuja: disease_status). Taulukosta valitaan arviointihetkellä oleva paras vaste.
|
Lyhenne |
Nimike |
Määritelmä |
|
CR-MRDneg |
Remissio, jäännostauti negatiivinen |
|
|
CR-MRDpos |
Remissio, jäännostauti positiivinen |
|
|
CCR |
Sytogeneettinen remissio |
|
|
CR |
Morfologinen remissio |
|
|
CRi |
Remissio, puutteellinen luuytimen toipuminen |
|
|
PR |
Osittainen remissio |
|
|
Hypoplasia |
Dysplasia/hypoplasia |
|
|
RD |
Refraktaari tauti (RD) |
|
|
Relapsi (MRD) |
Molekyyligenettinen relapsi |
|
|
Relapsi (bm) |
Relapsi |
|
|
Relapsi (neuro) |
Isololoitu neurorelapsi |
|
|
Relapsi (ekstram) |
Isololoitu ekstramedullaarinen relapsi |
|
| Exitus- refraktaari | Exitus, refraktaari tauti |
|
|
Exitus-relapsi |
Exitus, relapsi |
|
|
Exitus-infektio |
Exitus, infektio |
|
|
Exitus-aplasia |
Exitus, aplasia |
|
|
Exitus-hoitoon liittyvä (TRM) |
Exitus, hoitoon liittyvä |
|
|
Exitus-muu tauti |
Exitus toisesta taudista johtuen |
|
| Exitus - ei hoitoa | Exitus, hoito keskeytetty |
|
| Exitus - ei edeltävää hoitoa |
Exitus, palliatiivinen hoito |
|
|
Exitus-tuntematon |
Exitus, syy tuntematon |
|
|
Poistunut seurannasta |
Seuranta loppunut (lost-to-follow-up) |
|
Hoito
Hoito
Sivusto siirtymässä toiseen osoitteseen. Ajankohtaiset ohjeet löytyvät toistaiseksi linkistä: https://www.hematology.fi/fi/hoito-ohjeet/veritaudit/akuutit-leukemiat/a...
AML-potilaat hoidetaan ensisijaisesti SLR:n AML-2018-hoito-ohjelman mukaisesti. Akuutti promyelosyyttileukemia (APL) hoidetaan erillisen ohjeen mukaisesti.
AML-2012 (vanha protokolla)
- Kirjaudu tai rekisteröidy kommentoidaksesi
AML-2012 (vanha protokolla)
Täältä löydät tietoa Suomen leukemiaryhmän AML-2012-hoito-ohjelmasta (lopullisen version päiväys 11.01.2012). Se on kokonaisuudessaan ladattavissa alla olevasta liitetiedostosta (attachment). Alla otsikoittain jaoteltuna on esitetty keskeiset osiot hoito-ohjelmasta.
| Liite | Koko |
|---|---|
| 293 KB |
Tiivistelmä AML-2012
- Kirjaudu tai rekisteröidy kommentoidaksesi
Tiivistelmä AML-2012
Kyseessä on 16-vuotiaille ja sitä vanhemmille akuuttia myelooista leukemiaa (AML) sairastaville potilaille tarkoitettu valtakunnallinen Suomen leukemiaryhmän (SLR) laatima hoito-ohjelma, jonka tarkoitus on saada kaikki yli 65-vuotiaat potilaat yhtenäisen hoidon piiriin.
Hoito-ohjelmaan soveltuva potilas saa aluksi induktiohoidon (enimmillään kaksi sykliä), jolla tähdätään morfologiseen remissioon. Remissio pyritään mahdollisuuksien mukaan arvioimaan luuydinnäytteestä myös virtaussytometrisellä menetelmällä. Morfologisen remission saavuttaneet potilaat jaetaan kolmeen eri riskiluokkaan (pieni riski, keskiriski ja suuri riski) dg-vaiheessa otettujen geneettisten tutkimustulosten mukaisesti. Jokaisessa riskiluokassa toteutetaan remission jälkeen konsolidaationa enimmillään kolme solunsalpaajahoitoa, jonka jälkeen siirrytään seurantaan. Kuitenkin kaikille niille remission saavuttaneille allogeeniseen siirtoon soveltuville keskiriskin ja suuren riskin potilaille, joille sopiva luovuttaja löytyy, suositellaan ensimmäisessä remissiossa konsolidaationa allogeenista kantasolusiirtohoitoa heti kun se on mahdollista toteuttaa. Pienen riskin potilaille suositellaan allogeenista kantasolusiirtohoitoa ensimmäisessä remissiossa vain, jos kolmen konsolidaatiohoidon jälkeen heillä on molekulaarisesti mitattava merkittävä jäännöstauti.
Jäännöstautia seurataan allogeeniseen kantasolusiirtohoitoon soveltuvilla geneettisesti pienen riskin potilailla jokaisen hoidon jälkeen ja seurantavaiheessa kolmen kuukauden välein kahteen vuoteen saakka remission toteamisesta. Jos tällaiselle potilaalle ilmaantuu seurannan aikana merkittävä jäännöstauti, hänelle suositellaan allogeenista kantasolusiirtohoitoa, jos se on mahdollista toteuttaa. Muissa riskiryhmissä jäännöstaudin seuranta ei ole välttämätön, mutta sitä suositellaan tehtäväksi kuten pienen riskin ryhmässä, tutkimustiedon kartuttamiseksi tulevia hoitotutkimuksia ajatellen. Pienen riskin potilaille jäännöstaudin seurantaan soveltuvin menetelmä on RQ-PCR. Muissa riskiryhmissä jäännöstaudin seurantaan käytetään soveltuvinta herkintä menetelmää. Jäännöstaudin seurantaa suositellaan tehtäväksi myös virtaussytometrialla.
Induktiohoidolle refraktaaristen ja hoidon tai seurannan aikana relaboituvien potilaiden jatkohoidosta päättää hoitava lääkäri.
Potilaat ja hoitotulosten arvioimisessa tarvittavat seurantatiedot (erillinen raportointiliite) ilmoitetaan potilaan antamalla kirjallisella suostumuksella Suomen Hematologiseen Rekisteriin (SHR). Jokainen vastuulääkäri ohjeistaa ERVA-alueensa keskussairaalat hoito-ohjelman ja sen raportoinnin suhteen.
Hoito-ohjelmassa mukana oleville potilaille suositellaan osallistumista mahdollisesti myöhemmin käynnistyviin erillisiin SLR:n suunnittelemiin AML:n tutkimuksiin.
Hoito-ohjelmaan pyritään saamaan 400 potilasta vuosien 2012 - 2015 aikana.
Vuokaaviot (AML-2012)
- Kirjaudu tai rekisteröidy kommentoidaksesi
Vuokaaviot (AML-2012)
AML-2012-hoito-ohjelma: yleiskaavio
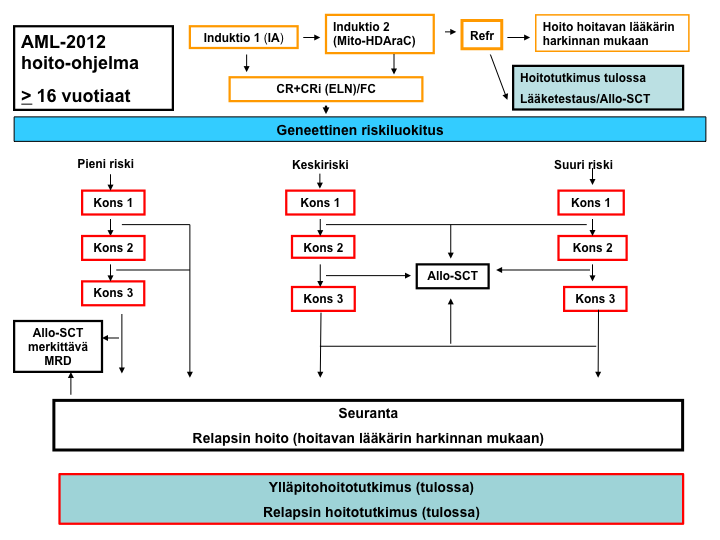
Pienen relapsiriskin hoitokaavio
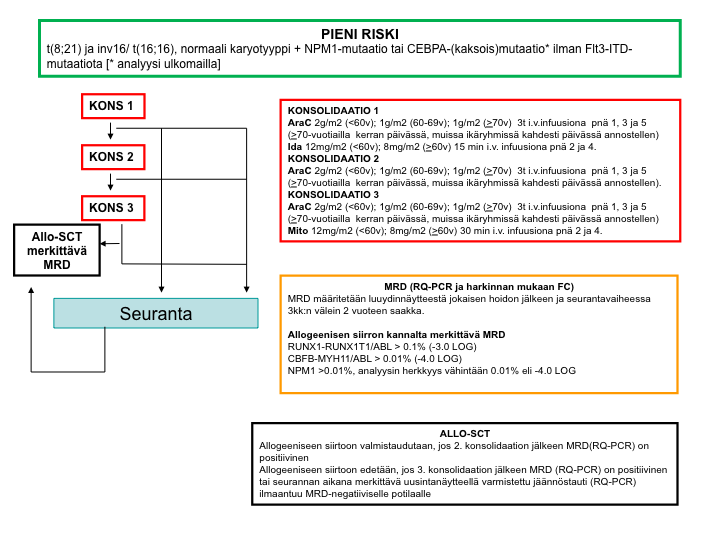
Keskisuuren relapsiriskin hoitokaavio
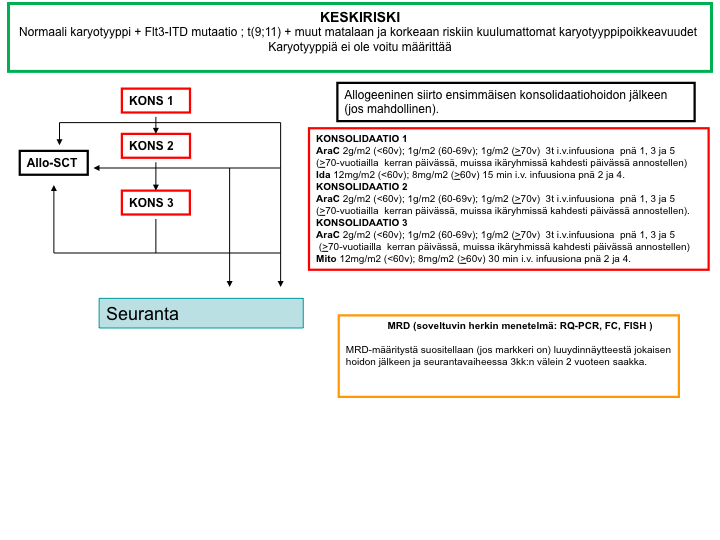
Suuren relapsiriskin hoitokaavio
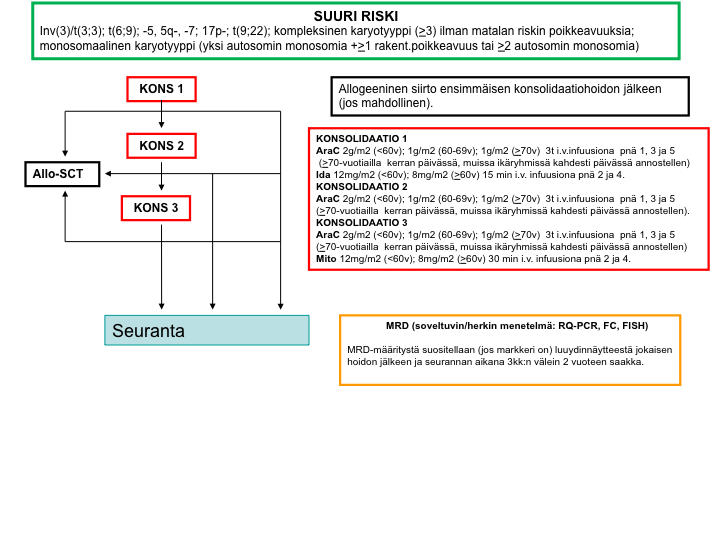
Valintakriteerit
- Kirjaudu tai rekisteröidy kommentoidaksesi
Valintakriteerit
- Potilaan ikä on dg-hetkellä >16 vuotta
- Potilaalla on AML WHO-kriteerien mukaisesti
- Allekirjoitettu kirjallinen suostumus tietojen tallentamisesta Suomen Hematologiseen Rekisteriin (FHRB)
Poissulkukriteerit
- Akuutti promyelosyyttileukemia
- Soveltumattomuus hoito-ohjelmaan: vaikea sydän-, keuhko-, maksa- tai munuaissairaus, vaikea psykiatrinen sairaus/tila, vaikea dementia, huono hoitomyöntyvyys
- Arvioitu elinaika leukemiasta riippumattomasta syystä on alle 6 kk.
Hoito-ohjelman ensisijaiset päätetapahtumat
- Remissio (CR)
- Relapsivapaa elinaika (RFS)
- Kumulatiivinen relapsin insidenssi (CIR)
- Kokonaiselinaika
Hoito-ohjelman toissijaiset päätetapahtumat
- Induktio- ja konsolidaatiohoidon kuolleisuus
- Hoitosyklin viivästymisen syyt
- Hoito-ohjelman keskeyttämisen syyt
Muita raportoitavia parametreja
Jokaisesta hoitosyklistä:
- Neutropenian kesto (<0.5)
- Trombosytopenian kesto (<50)
- Infektioiden vaikeusaste (WHO-luokitus)
- Veriviljelypositiiviset infektiot
- Sairaalapäivät
- Jäännöstaudin määrä
Seurantavaihe:
- Jäännöstaudin määrä 3 kk välein 2 vuoteen saakka
Induktiohoidot (AML-2012)
- Kirjaudu tai rekisteröidy kommentoidaksesi
Induktiohoidot (AML-2012)
Ensimmäinen induktio (IA)
Idarubisiini (Ida): 12 mg/m2 päivinä 1, 3 ja 5 15 min iv infuusiona (sama annos kaikissa ikäryhmissä)
Sytarabiini (AraC):
• <60-vuotiaat: bolus 100 mg päivänä 1 ja 100 mg/m2 jatkuvana iv infuusiona pnä 1-9
• ≥60-vuotiaat: bolus 100 mg päivänä 1 ja 100 mg/m2 jatkuvana iv infuusiona pnä 1-7
Toinen induktio (Mito-HDAraC)
Se annetaan vain, jos potilas ei mene morfologiseen remissioon ensimmäisellä induktiolla.
Mitoksantroni (Mito):
• <60-vuotiaat: 12 mg/m2 30 min iv. infuusiona pnä 2-5
• ≥60-vuotiaat: 8 mg/m2 30 min iv. infuusiona pnä 2-5
Suuriannos-sytarabiini (HDAraC):
• <60-vuotiaat: 2 g/m2 3 tunnin iv. infuusiona 12 tunnin välein päivinä 1, 3, 5 ja 7
• 60-69-vuotiaat: 1 g/m2 3 tunnin iv. infuusiona 12 tunnin välein päivinä 1,3, 5 ja 7
• ≥70-vuotiaat: 1 g/m2 3 tunnin iv. infuusiona kerran päivässä päivinä 1, 3, 5 ja 7
• Oftan Dexa silmätipat 1gttx4-6 molempiin silmiin pnä 1-8
Konsolidaatiohoidot (AML-2012)
- Kirjaudu tai rekisteröidy kommentoidaksesi
Konsolidaatiohoidot (AML-2012)
Ensimmäinen konsolidaatiohoito (HDAraC-Ida)
Annetaan vain, jos potilas saavuttaa morfologisen remission joko ensimmäisellä tai toisella induktiohoidolla.
Suuriannos-sytarabiini (HDAraC):
• <60-vuotiaat: 2 g/m2 3 tunnin iv. infuusiona 12 tunnin välein pnä 1, 3 ja 5
• 60-69-vuotiaat: 1 g/m2 3 tunnin iv infuusiona 12 tunnin välein pnä 1, 3 ja 5
• ≥70-vuotiaat: 1 g/m2 3 tunnin iv. infuusiona kerran päivässä pnä 1, 3 ja 5
• deksametasoni-silmätipat 1 gtt x4-6 molempiin silmiin pnä 1-6
Idarubisiini:
• <60-vuotiaat: 12 mg/m2 15 min iv. infuusiona pnä 2 ja 4
• ≥60-vuotiaat: 8 mg/m2 15 min iv. infuusiona pnä 2 ja 4
Toinen konsolidaatiohoito (HDAraC)
Se annetaan vain, jos potilas on morfologisessa remissiossa.
Suuriannos-sytarabiini (HDAraC):
• <60-vuotiaat: 2 g/m2 3 tunnin iv. infuusiona 12 tunnin välein pnä 1, 3 ja 5
• 60-69-vuotiaat: 1 g/m2 3 tunnin iv infuusiona 12 tunnin välein pnä 1, 3 ja 5
• ≥70-vuotiaat: 1 g/m2 3 tunnin iv. infuusiona kerran päivässä pnä 1, 3 ja 5
• deksametasoni-silmätipat 1 gtt x4-6 molempiin silmiin pnä 1-6
Kolmas konsolidaatiohoito (HDAraC-Mito)
Se annetaan vain, jos potilas on morfologisessa remissiossa.
Suuriannos-sytarabiini (HDAraC):
• <60-vuotiaat: 2 g/m2 3 tunnin iv. infuusiona 12 tunnin välein pnä 1, 3 ja 5
• 60-69-vuotiaat: 1 g/m2 3 tunnin iv infuusiona 12 tunnin välein pnä 1, 3 ja 5
• ≥70-vuotiaat: 1 g/m2 3 tunnin iv. infuusiona kerran päivässä pnä 1, 3 ja 5
• deksametasoni-silmätipat 1 gtt x4-6 molempiin silmiin pnä 1-6
Mitoksantroni (Mito):
• <60-vuotiaat: 12 mg/m2 30 min iv. infuusiona pnä 2 ja 4
• ≥60-vuotiaat: 8 mg/m2 30 min iv. infuusiona pnä 2 ja 4
Kantasolujensiirto (kriteerit AML-2012)
- Kirjaudu tai rekisteröidy kommentoidaksesi
Kantasolujensiirto (kriteerit AML-2012)
Pienen riskin AML-potilailla valmistaudutaan kolmannen hoidon jälkeen tehtävään allogeeniseen kantasolusiirtohoitoon, jos aikaisempien hoitojen jäännöstaudin perusteella se näyttää todennäköiseltä. Jos siirto ei ole mahdollinen, siirrytään seurantavaiheeseen.
Keskisuuren ja suuren riskin ryhmässä tehdään allogeeninen kantasolujensiirto kahden hoidon jälkeen tai sen jälkeen heti kun se on mahdollista, jos sopiva luovuttaja (sisarus tai rekisteristä peräisin oleva) on olemassa ja siirto muutoin on mahdollista toteuttaa. Jos luovuttajaa ei ole, annetaan potilaille yhteensä kolme konsolidaatiota. Kolmen konsolidaatiohoidon jälkeen tai konsolidaatiohoitojen keskeytyessä siirrytään seurantavaiheeseen.
Allogeenista kantasolujensiirtohoitoa tarjotaan edellä esitetyin perustein siirtoon soveltuvalle potilaalle 65 ikävuoteen saakka ja yksilöllistä harkintaa käyttäen aina 70 ikävuoteen saakka, jos hänelle löytyy sopiva luovuttaja. Siirtoon soveltuvista keskiriskin ja suuren riskin potilaista laaditaan lähete allogeeniseen siirtoyksikköön (HYKS tai TYKS) mahdollisimman varhaisessa vaiheessa. Myös pienen riskin potilaiden siirtoon varaudutaan mahdollisimman varhain jäännöstaudin tulos huomioiden. Lähetteeseen liitetään liitteen 7 mukaiset tiedot (ks. erillinen linkki ja protokolla). Jos sisarusluovuttajaa ei löydy, siirtokeskus ryhtyy etsimään rekisteriluovuttajaa. Samanaikaisesti tiiviisti seurataan potilaan hoidon etenemistä ja mahdollisten komplikaatioiden syntymistä. Jos potilaalle löytyy sisarusluovuttaja tai rekisteristä löytyy kudostyypiltään sopiva luovuttaja (siirtokeskuksen tekemä valinta), käydään siirtokeskuksessa keskustelu allogeeniseen siirtoon liittyvistä asioista potilaan, lähiomaisen ja mahdollisen sisarusluovuttajan kesken. Kun potilas on ilmoittanut keskustelun jälkeen haluavansa edetä siirtoon (ja sisarusluovuttaja samoin), voidaan tautitilanteen salliessa ryhtyä aktivoimaan siirtoaikataulua. Noin kolme viikkoa ennen kantasolujen siirtopäivää potilas ja sisarusluovuttaja tutkitaan polikliinisesti siirtoyksikössä. Mikäli tutkimuksissa ei ilmene estettä, edetään suunniteltuun intensiivihoitoon. Rekisterin luovuttaja tutkitaan rekisterin toimesta. Lopullisen arvion potilaan sopivuudesta siirtoon, luovuttajan valinnan ja potilaalle annettavan esihoidon tekee siirtoyksikkö.
Keskiriskin ja suuren riskin potilailla siirtoon pääsyn optimaalisin ajankohta on induktion tai yhden konsolidaation jälkeen.
Tietojen rekisteröinti
- Kirjaudu tai rekisteröidy kommentoidaksesi
Tietojen rekisteröinti
Hoito-ohjelman tiedot ovat SLR:n omaisuutta. Raportoitavat tiedot siirretään potilaan kirjallisella suostumuksella Suomen Hematologiseen Rekisteriin (SHR/FHRB). Raportoitavat tiedot voidaan siirtää joko suoraan rekisteriin tai ne voidaan ensin kerätä erillisille kaavakkeille ja niistä myöhemmin siirtää rekisteriin. Tietojen siirtäminen rekisteriin edellyttää sitä, että SHR on hyväksytty sairaanhoitopiireissä käyttöön (eettisen toimikunnan kannanotto ja shp:n johtajaylilääkärin hyväksyntä). Jokaisen ERVA-alueen vastuulääkäri huolehtii siitä, että SHR on saanut hyväksynnän alueellaan. Hän myös ohjeistaa tietojen keräämisen vastuualueellaan. Kerättyjä tietoja käsitellään salassa pidettävien tietojen vaatimusten mukaisesti. Vain nimettyjen henkilöiden on mahdollista tarkastella hoitotuloksia. SHR:n tiedoista tehdyistä yhteenvedoissa ja raporteissa yksittäisten potilaiden tietoja ei voida tunnistaa.
Allogeenisen kantasolusiirtohoidon saaneet potilaat raportoidaan tästä riippumatta transplantaatiorekistereihin normaalin käytännön mukaisesti ja tuloksia voidaan käyttää rekisterien tekemissä analyyseissä. Hoito-ohjelman tuloksista raportoidaan leukemiaryhmälle ja ne hyödynnetään niissä erillisissä hoitotutkimuksissa, joihin potilaita tästä hoito-ohjelmasta mahdollisesti suositellaan. Hoitotulokset pyritään myös raportoimaan alan kansainvälisissä lehdissä SLR:n toimesta. SLR päättää julkaisujen kirjoittajista erikseen.
Vastuulääkärit
- Kirjaudu tai rekisteröidy kommentoidaksesi
Vastuulääkärit
Pirjo Koistinen
AML-2012-hoito-ohjelman vastuuhenkilö
Syöpätaudit ja hematologia, Medisiininen tulosalue, OYS
PL 20, 90029 OYS
pirjo.koistinen@ppshp.fi, puh (08) 315 4617
Kimmo Porkka
Hematologian klinikka, Medisiininen tulosyksikkö, HYKS
PL 372, Haartmaninkatu 4, 00029 HUS
kimmo.porkka@helsinki.fi, puh. 09 471 72338
Tapio Nousiainen
Sisätautien klinikka, KYS
PL 1777, 70211 Kuopio
tapio.nousiainen@kuh.fi, puh. (017) 173 311
Maija Itälä-Remes
osasto 015, TYKS
PL52, 20521 Turku
maija.itala-remes@tyks.fi, puh (02) 313 0000
Hannele Rintala
Sisätautien klinikka, TAYS
PL 2000, 33521 Tampere
hannele.rintala@pshp.fi, puh (03) 311 611
Potilaan tiedote
- Kirjaudu tai rekisteröidy kommentoidaksesi
Potilaan tiedote
Versio 17.04.2012
Tietoa akuutista myelooisesta leukemiasta ja sen uudesta suomalaisesta hoito-ohjelmasta
16-VUOTIAIDEN JA SITÄ VANHEMPIEN AKUUTTIA MYELOOISTA LEUKEMIAA SAIRASTAVIEN POTILAIDEN HOITO-OHJELMA
Lyhenne: AML-2012
Arvoisa potilas
Tässä tiedotteessa kerrotaan akuutin myelooisen leukemian uudesta hoito-ohjelmasta, jonka piiriin pyritään saamaan mahdollisimman moni kyseiseen leukemiaan sairastuva aikuispotilas Suomessa. Hoito-ohjelma on Suomen Leukemiaryhmän laatima ja siinä hyödynnetään viimeaikaisin tutkimustieto taudista ja sen hoidosta.
Akuutin myelooisen leukemian hoidon yleisiä periaatteita
Akuutti myelooinen leukemia on aikuisten yleisin akuutti leukemia, jossa luuydin tuottaa epäkypsiä blastisoluja (leukemiasoluja) verenkiertoon, josta ne voivat levitä muualle elimistöön. Samanaikaisesti luuytimen normaali verisolumuodostus estyy ja aiheuttaa anemian, terveiden valkosolujen vähyyden ja verihiutaleiden kadon. Terveiden valkosolujen vähyys johtaa toistuviin infektioihin ja verihiutaleiden eli trombosyyttien kato lisää verenvuotoja. Veren suuri blastimäärä voi johtaa verenkiertohäiriöihin ja eri elinten normaalin toiminnan häiriintymiseen. Hoitamattomana tauti johtaa nopeasti kuolemaan.
Akuutin myelooisen leukemian hoidon kulmakivi on suuriannoksinen solunsalpaajalääkitys, joita annetaan muutaman päivän kuureina 4 – 6 viikon välein. Yleensä hoitokuureja tarvitaan useita. Hoidon päämääränä on taudista paraneminen, johon päästään tuhoamalla kaikki elimistössä olevat leukemiasolut.
Leukemian hoidot aiheuttavat veren kaikkien soluarvojen voimakkaan mutta ohimenevän laskun, mikä selittää suuren osan hoitoon liittyvistä haittavaikutuksista. Haittavaikutuksina voi esiintyä väsymystä, tulehduksia ja verenvuotoja. Suun kipeytyminen, ruokahaluttomuus, pahoinvointi, ripuli ja hiusten lähtö ovat myös tavallisia. Mahdollisuus lasten saamiseen huononee hoitojen takia.
Suurin osa haittavaikutuksista on kuitenkin pääsääntöisesti hoidettavissa tukihoidoin. Tukihoitoina tarvitaan verituotteita ja antibiootteja, joita annetaan toistuvasti laskimonsisäisesti. Hoitojen aikana joudutaan turvautumaan usein myös suonensisäiseen ravitsemushoitoon. Hoitojen helpottamiseksi tarvitaan lähes poikkeuksetta keskuslaskimokatetria, jonka anestesialääkäri laittaa paikallispuudutuksen avulla suureen laskimoon. Katetri on tarkoitettu pitkäaikaiseen käyttöön, eikä se estä esimerkiksi kotona käymistä hoitojen välissä.
Ensimmäisellä hoitojaksolla pyritään saavuttamaan remissio, jolloin leukemiasoluja ei enää nähdä luuytimessä, veriarvot ovat normaalistuneet ja leukemian mahdollisesti aiheuttamat oireet ovat hävinneet. Myöhempien hoitojaksojen tarkoituksena on hävittää mahdollinen mikroskoopilla havaitsematon jäännöstauti ja estää taudin uusiutuminen eli relapsi.
Normaalin käytännön mukaisesti hoidon tuloksia seurataan veri- ja luuydinnäytteiden avulla. Verinäytteitä otetaan voimakkaiden hoitojen jälkeen lähes päivittäin. Luuydinnäytteitä tutkitaan noin kuukauden kuluttua ensimmäisen hoidon alusta ja myöhemmin hoitokuurien jälkeen veriarvojen toivuttua sekä seurantavaiheessa neljä kertaa vuodessa kahden vuoden ajan. Kantasolujensiirron hyödyistä ja riskeistä järjestetään erillinen keskustelu niille potilaille, jotka ovat siihen soveliaita ja joille tätä hoitomuotoa tarjotaan 1 – 3 solunsalpaajakuurin jälkeen.
Hoitokuurit annetaan yleensä sairaalassa. Infektioiden ja verenvuotojen vaaran takia joudutaan hoitokuurien jälkeen olemaan sairaalassa yleensä muutamia viikkoja. Sairaalahoitojaksojen välillä lyhyet kotilomat ovat usein mahdollisia.
Hoito-ohjelman suunnittelija – Suomen Leukemiaryhmä
Maassamme akuuttien leukemioiden hoito ohjelmat on suunniteltu yli 25 vuoden ajan Suomen Leukemiaryhmän toimesta. Ryhmään kuuluu lääkäreitä pahanlaatuisia veritauteja hoitavista sairaaloista. Yhteisen suunnittelun ja tulosten seuraamisen tarkoituksena on kehittää Suomen oloihin parhaiten soveltuvia ja tehokkaita hoito ohjelmia. Käsillä oleva hoito-ohjelma toteutetaan kaikissa Suomen yliopistollisissa sairaaloissa ja joissakin keskussairaaloissa. Tähän mennessä toteutetuilla hoito- ja tutkimusohjelmilla saavutetut hoitotulokset ovat parantuneet koko ajan ja ovat myös kansainvälisesti arvioituna korkeatasoisia.
Miksi tämä hoito-ohjelma on laadittu?
Leukemian hoidossa käytetään monenlaisia solunsalpaajahoitojen yhdistelmiä, joilla on saavutettu kohtalaisen hyviä hoitotuloksia. Tällä hetkellä ei voida sanoa mikä yhdistelmistä on kaikkein paras. Tämän takia yksityiskohdiltaan toisistaan eroavia hoito-ohjelmia on syytä edelleen arvioida niin tehon kuin haittavaikutustenkin suhteen.
Tässä hoito-ohjelmassa kaikki potilaat saavat saman remissioon tähtäävän induktiohoidon sekä remission saavuttamisen jälkeen kolme konsolidaatiohoitoa taudin uusiutumisen estämiseksi. Potilaat jaetaan tiettyjen diagnoosivaiheessa todettujen riskitekijöiden perusteella kolmeen eri riskiluokkaan, mikä vaikuttaa siihen, suositellaanko etenemistä allogeeniseen kantasolusiirtoon, jos sopiva kantasolujen luovuttaja löytyy.
Akuutin myelooisen leukemian ennuste vaihtelee potilaittain huomattavasti. Diagnoosivaiheen löydösten perusteella hoitovasteen saannin mahdollisuutta ja leukemian uusiutumisen riskiä voidaan arvioida monin tavoin. Viime vuosina sairaalakäyttöön on kehitetty aiempaa herkempiä menetelmiä elimistöön hoidon jälkeen mahdollisesti jäävien leukemiasolujen eli jäännöstaudin toteamiseksi. Herkät tutkimusmenetelmät ovat tuoneet runsaasti uutta tietoa leukemian hoitoon, mutta niihin liittyy myös runsaasti selvitettäviä kysymyksiä sekä laboratoriotekniikan että tulosten arvioinnin kannalta. Tässä hoito-ohjelmassa nämä riskitekijät kartoitetaan uusien kansainvälisten ohjeiden mukaisesti ja mahdollista jäännöstautilöydöstä pyritään seuraamaan luuydinnäytteestä säännöllisin väliajoin aina 2 vuoteen saakka sairastumisesta.
Hoito-ohjelman tavoite
Hoitojen jatkuvan kehittämisen takia on tärkeää kerätä tiedot paitsi hoitotuloksista myös leukemian luonteesta ja potilaskohtaisista hoidon tuloksiin vaikuttavista tekijöistä. Ohjelmaan pyritään saamaan kaikki Suomessa hoidettavat 16-vuotiaat ja sitä iäkkäämmät voimakkaaseen solunsalpaaja-hoitoon soveltuvat akuuttia myelooista leukemiaa sairastavat potilaat. Hoitoon soveltuvuuttaa arvioidaan aikaisempaa paremmin hyödyntäen WHO:n toimintakykymittaristoa sekä muun sairastavuuden pisteytystä siihen kehitetyillä ohjeistuksilla.
Hoito-ohjelman tärkeä tavoite on saada myös iäkkäät potilaat parantavan hoidon piiriin ja samalla arvioida, parantaako voimakas hoito iäkkäiden potilaiden ennustetta, ja onko hoidon toksisuus hyväksyttävissä. Alle 65-vuotiaiden kohdalla hoitotuloksia voidaan verrata historiallisiin kontrolleihin eli AML-92 ja AML-03-hoitotutkimuksiin osallistuneiden potilaiden hoitotuloksiin.
Tässä hoito-ohjelmassa potilaiden ohjaaminen allogeeniseen kantasolusiirtohoitoon nopeutuu ja tehostuu. Samalla kaikki (myös iäkkäät) yleisten suositusten mukaan allogeenisen kantasolusiirtohoidon piiriin kuuluvat potilaat saadaan systemaattisesti poimittua siirto-ohjelmaan.
Hoito-ohjelman yksi tavoite on arvioida jäännöstaudin merkitys kaikissa riskiryhmissä potilaan ennusteeseen. Tässä hoito-ohjelmassa tätä tietoa kerätään entistä systemaattisemmin ja useammalla eri menetelmällä. Tieto voidaan hyödyntää tulevaisuudessa arvioitaessa sitä, missä vaiheessa ja minkä suuruinen jäännöstauti tulisi hoitoa tehostamalla pyrkiä eliminoimaan.
Diagnoosi-, remissio- ja mahdollisessa relapsivaiheessa kerättävät veri- ja luuydinnäytteet, sekä kliinisen tiedon keräys Suomen hematologiseen rekisteriin ja biopankkiin (FHRB) mahdollistavat akuutin leukemian hoidon jatkuvan kehitystyön. Rekisteriin kerätty data helpottaa ja nopeuttaa hoito-ohjelman tulosten tilastollista analysointia ja väliraporttien tekemistä. Tästä tutkimuksesta saatte erillisen tiedotteen ja suostumuskaavakkeen.
Teiltä pyydetään kirjallinen suostumus biopankkinäytteiden ottamiseen sekä hoitotietojen siirtämiseen ja raportointiin Suomen Hematologiseen Rekisteriin. Mahdollinen kieltäytymisenne tietojenne rekisteröinnistä tai tutkimusnäytteiden otosta ei vaaranna tautinne hoitoa tämän hoito-ohjelman tai muun tunnetun hoito-ohjeistuksen mukaisesti. Kaikki hoito-ohjelman aikana kerätty tieto käsitellään luottamuksellisena eikä hoito-ohjelmaan osallistuvan henkilöllisyys käy ilmi tehtävissä yhteenvedoissa eikä raporteissa. Suomen Hematologista Rekisteriä, johon tiedot kootaan, säilytetään tietosuojalainsäädännön edellyttämällä tavalla ja sen käyttö on hyväksytty Teitä hoitavassa sairaalassa.
AML-2018
AML-2018
APL
- Kirjaudu tai rekisteröidy kommentoidaksesi
APL
Sivusto on siirtymässä toiseen osoitteseen. Akuutin promyelosyyttileukemian (APL) hoito-ohje (Suomen AML-ryhmä) alla, muut ohjeet ks https://www.hematology.fi/fi/hoito-ohjeet/veritaudit/akuutit-leukemiat/a...
| Liite | Koko |
|---|---|
| 526.32 KB |
Hoito-ohjelmia
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hoito-ohjelmia
AI-ETI
- Kirjaudu tai rekisteröidy kommentoidaksesi
AI-ETI
Suomen leukemiaryhmän AML96 ohjelman hoito. Hoito on verraten hyvin siedetty, mutta relapseja tulee runsaasti. Induktiohoitona on 6 päivän AI-hoito, minkä jälkeen annetaan kaksi ETI-hoitoa. Näiden jälkeen annetaan oraalista merkaptopuriini- metotreksaatti -ylläpitohoitoa kunnes kolme vuotta on kulunut diagnoosihetkestä tai kunnes ilmaantuu relapsi.
Käyttöaiheet
- Iäkkäiden AML-taudin hoito
AI-hoito 6 päivää
- Sytarabiini 100 mg/m2 x 2 15 minuutin infuusiona 12 tunnin välein päivinä 1-6
- Idarubisiini 12 mg/m2 10 minuutin infuusiona päivinä 4 ja 6 (2 annosta)
ETI‑hoito
- Etoposidi 80 mg/m2 x 2 po. päivinä 1‑5
- Tioguaniini 100 mg/m2 x 2 po. päivinä 1‑5
- Idarubisiini 15 mg/m2 x 1 po. päivinä 1‑3
Merkaptopuriini ‑ metotreksaatti ‑ ylläpitohoito
- Merkaptopuriini 70 mg/m2 kerran päivässä po.
- Metotreksaatti 15 mg/m2 kerran viikossa po.
Kirjallisuutta
Suomen leukemiaryhmän tutkimusprotokolla
Otsikko vaihdettiin muodosta <em class="placeholder">AI-ETI</em> muotoon <em class="placeholder">AI-ETI</em>.
AML2003
- Kirjaudu tai rekisteröidy kommentoidaksesi
AML2003
Sisäänottokriteerit
Ikä 16-65 v, AML WHO kriteerein, potilaan suostumus. Potilastiedotteita ja suostumuslomakkeita on osastoilla, pkl:lla ja tämän sivun alalaidassa liitteenä (attachment)
Poissulkukriteerit
APL, KMLbla, muu sairaus estää voimakkaan hoidon antamisen, raskaus, imetys.
Diagnoosivaiheen tutkimukset
Anamneesi
Selvitetään sekundaarileukemian mahdollisuus, onko potilas saanut säde- tai solunsalpaajahoitoa tai onko hän altistunut erityisen voimakkaasti luuydintä vaurioittaville kemikaaleille tai säteilylle. Selvitetään sisarusten olemassaolo ja heidän terveydentilansa karkeasti. Hoidon toteutumiseen vaikuttavat muut sairaudet, lääkitys ja allergiat.
Status
Kartoitetaan infektio- ja vuototilanne, etsitään taudin ekstramedullaarisia ilmentymiä (imusolmukealueet, perna ja maksa, iho, ikenet, rinnat, kivekset), arvioidaan vitaalielinten tila ja potilaan suorituskyky.
Laboratoriotutkimukset
Akuutin leukemian alkukokeet edellä olevan mukaisesti.
Muut tutkimukset
Vatsan UÄ: pernan ja maksan koko ja rakenne. Thorax-röntgenkuva. Infektioiden kartoitus tilanteen mukaan. EKG. Muut tutkimukset tilanteen mukaan.
Potilaan sisarusten kudostyypitykset
On tärkeää järjestää alle 61-vuotiaiden mahdollisesti allogeniseen siirtoon soveltuvien potilaiden sisarusten HLA-tyypitys nopeasti! Luovuttajaehdokkailla ei saa olla sellaisia vaikeita sairauksia, jotka aiheuttaisivat siirteen luovutuksen yhteydessä riskejä.
Induktio- ja konsolidaatiohoitojen aikaiset tutkimukset
Luuydinnäytteet
Luuytimestä tutkitaan induktiohoidon ja jokaisen konsolidaatiohoidon jälkeen
- Morfologia
- Valittu jäännöstaudin markkeri, jos sellainen on diagnoosivaiheessa löydetty. Jos markkeri tutkitaan kromosomilaboratoriossa, otetaan lisäksi näytettä 7-10 ml kahteen CPT-putkeen mahdollisia myöhemmin tehtäviä täydentäviä tutkimuksia varten (blancho 99999).
- Mononukleaaristen solujen eristys ja mRNA:n eristys kantasolulaboratoriossa (Bm-SoluHem 19465).
Hoitojaksojen yhteydessä arvioidaan:
Neutropenian kesto (< 0,5 x 109/l), trombosytopenian kesto (< 50 x 109/l), korkein CRP, kuumepäivien lukumäärä (³38 oC), sairaalapäivien lukumäärä, huonoin suoriutumiskyky , infektion vaikeusaste, veriviljely-positiiviset infektiot, suolitoksisuus, maksatoksisuus, CNS-toksisuus, ihotoksisuus, muu mahdollinen toksisuus, punasolusiirtojen ja trombosyyttisiirtojen tarve.
Kantasolujen luovuttajan hakeminen
HLA II -näyte sisarus- tai rekisteriluovuttajan hakua varten tutkitaan, jos siirto on aiheellinen riskiryhmäluokituksen ja jos se on mahdollinen allogeenisen siirron edellytysten perusteella. Näyte tutkitaan siirtoikäisiltä potilailta viimeistään ennen toista hoitoa.
Optimaalisen hoitotuloksen kannalta on tärkeää selvittää luovuttajan olemassaolo mahdollisimman nopeasti diagnoosivaiheessa. Ainakin alustavasti tiedon tulisi olla käytettävissä sisarusluovuttajan osalta päivään 28 mennessä (1. HLA-tyypityksen tulos) ja rekisteriluovuttajan osalta päivään 56 mennessä (mahdollisesti sopivia ehdokkaita rekisterissä).
Seurantavaiheen tutkimukset konsolidaatiohoidon päätyttyä
1.- 2. vuosikonsolidaatiohoidon päättymisestä
- täydellinen verenkuva 1 kk välein.
- luuytimen aspiraationäyte 3 kk välein:
- morfologia
- valittu residuaalitaudin markkeri,
- mononukleaaristen solujen eristys ja mRNA:n eristys kantasolulaboratoriossa (Bm-SoluHem 19465).
3. - 5. vuosikonsolidaatiohoidon päättymisestä
- täydellinen verenkuva 4 kk välein.
- 3. vuodesta alkaen luuydin tutkitaan relapsiepäilyn herätessä.
6. - 10. vuosikonsolidaatiohoidon päättymisestä
- täydellinen verenkuva 6 kk välein.
- luuydin tutkitaan relapsiepäilyn herätessä.
| Liite | Koko |
|---|---|
| 67.5 KB | |
| 29.5 KB |
Induktiohoidot
- Kirjaudu tai rekisteröidy kommentoidaksesi
Induktiohoidot
Huom!: Randomisaatio IAT tai IdAraC-Ida-hoitoon
1. induktiohoito, randomoitu IAT-hoitoon
Alle 60-vuotiaat potilaat, 9 pv hoito
- Idarubisiini 12 mg/m2 päivinä 1, 3 ja 5, 15 minuutin infuusiona, yhteensä 3 annosta.
- Sytarabiini 50 mg/m2 15 min infuusiona päivänä 1 ja jatkuvana infuusiona 100 mg/m2/vrk päivinä 1 - 9, yhteensä koko hoidossa 950 mg/m2.
- Tioguaniini 75 mg/m2 suun kautta 12 tunnin välein päivinä 1 - 9, yhteensä 18 annosta.
- Sytarabiini- ja tioguaniinihoidon kesto lyhennetään 7 päiväksi, jos potilaalla on vaikea infektio kuten sepsis, lohkokeuhkokuume tms.
|
Lääke |
Annos mg/m2 |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Idarubisiini |
12 |
15 min inf |
x 1 |
1, 3 ja 5 |
|
Sytarabiini |
50 |
nopea inf |
x 1 |
1 |
|
Sytarabiini |
100 |
24 t inf |
jatkuva inf |
1 - 9 |
|
75 |
po. |
x 2 |
1 - 9 |
60 vuotta täyttäneet potilaat, 7 pv hoito
- Idarubisiini 12 mg/m2 päivinä 1, 3 ja 5, 15 minuutin infuusiona, yhteensä 3 annosta.
- Sytarabiini 50 mg/m2 15 min infuusiona päivänä 1 ja jatkuvana infuusiona 100 mg/m2/vrk päivinä 1 - 7, yhteensä koko hoidossa 750 mg/m2.
- Tioguaniini 75 mg/m2 suun kautta 12 tunnin välein päivinä 1 - 7, yhteensä 14 annosta.
|
Lääke |
Annos mg/m2 |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Idarubisiini |
12 |
15 min inf |
x 1 |
1, 3 ja 5 |
|
Sytarabiini |
50 |
nopea inf |
x 1 |
1 |
|
Sytarabiini |
100 |
24 t inf |
jatkuva inf |
1 - 7 |
|
Tioguaniini |
75 |
po. |
x 2 |
1 - 7 |
1. induktiohoito, randomoitu IdAraC-Ida-hoitoon
Alle 60-vuotiaat potilaat, 7 pv hoito
- Sytarabiini 1000 mg/m2 3 tunnin infuusiona x 2 päivinä 1 - 4, yhteensä 8 annosta.
- Idarubisiini 12 mg/m2 15 minuutin infuusiona päivinä 5 - 7, yhteensä 3 annosta.
- Kortikosteroidisilmätipat 1 gtt x 4 kumpaankin silmään päivinä 1 - 4.
|
Lääke |
Annos |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Sytarabiini |
1000 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Idarubisiini |
12 mg/m2 |
15 min inf |
x 1 |
5 - 7 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
60 vuotta täyttäneet potilaat, 6 pv hoito
- Sytarabiini 750 mg/m2 3 tunnin infuusiona x 2 päivinä 1 - 4, yhteensä 8 annosta.
- Idarubisiini 12 mg/m2 infuusiona päivinä 4 – 6, yhteensä 3 annosta.
- Kortikosteroidisilmätipat 1 gtt x 4 kumpaankin silmään päivinä 1 - 4.
|
Lääke |
Annos |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Sytarabiini |
750 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Idarubisiini |
12 mg/m2 |
15 min inf |
x 1 |
4 - 6 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
2. induktiohoito, MEA +/- GO
Toinen induktiohoito annetaan vain niille potilaille, joilla 1. induktiohoidon jälkeen blastien osuus normosellulaarisessa tai hypersellulaarisessa luuytimessä on yli 10 %.
Alle 60-vuotiaat potilaat
- Mitoksantroni 12 mg/m2 30 min iv. infuusiona kerran päivässä päivinä 2 - 5, yhteensä 4 annosta.
- Etoposidi (etoposidifosfaattina) 100 mg/m2 20 minuutin infuusiona kerran päivässä päivinä 1 - 4, yhteensä 4 annosta.
- Sytarabiini 1000 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
- Gemtutsumabi otsogamisiini (GO) potilaan kehon pinta-alan mukaan 5 mg (pinta-ala alle 1,7 m2) tai 10 mg (pinta-ala vähintään 1,7 m2) 2 tunnin infuusiona päivänä 1. GO laimennetaan 100 ml:aan 0,9 % NaCl. GO ei anneta niille potilaille, joille allogeeninen kantasolujensiirto on mahdollinen hoitomuoto.
|
Lääke |
Annos |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Mitoksantroni |
12 mg/m2 |
30 min inf |
x 1 |
2 - 5 |
|
Etoposidi |
100 mg/m2 |
20 min inf |
x 1 |
1 - 4 |
|
Sytarabiini |
1000 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
|
Gemtutsumabi otsogamisiini (MylotargR) jos ei allosiirtoa |
5 mg jos pinta-ala on alle 1,7 m2 10 mg jos pinta-ala on vähintään 1,7 m2 |
2 t inf |
x 1 |
1 |
60 vuotta täyttäneet potilaat
- Mitoksantroni 8 mg/m2 30 min infuusiona kerran päivässä päivinä 2 – 5, yhteensä 4 annosta.
- Etoposidi (etoposidifosfaattina) 100 mg/m2 20 min infuusiona kerran päivässä päivinä 1 – 4, yhteensä 4 annosta.
- Sytarabiini 500 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
- Gemtutsumabi otsogamisiini (GO) potilaan kehon pinta-alan mukaan 5 mg (pinta-ala alle 1,7 m2) tai 10 mg (pinta-ala vähintään 1,7 m2) 2 tunnin infuusiona päivänä 1. GO laimennetaan 100 ml:aan 0,9 % NaCl.
|
Lääke |
Annos |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Mitoksantroni |
8 mg/m2 |
30 min inf |
x 1 |
2 - 5 |
|
Etoposidi |
100 mg/m2 |
20 min inf |
x 1 |
1 - 4 |
|
Sytarabiini |
500 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
|
Gemtutsumabi otsogamisiini (MylotargR) |
5 mg jos pinta-ala on alle 1,7 m2 10 mg jos pinta-ala on vähintään 1,7 m2 |
2 t inf |
x 1 |
1 |
Siirtyminen konsolidaatiohoitoon
Jos 1. tai 2. induktiohoidon jälkeen luuytimen blastien osuus on 10 % tai vähemmän, siirrytään konsolidaatiohoito-ohjelmaan. Jos 2. induktiohoidon jälkeen luuytimen blastien osuus on yli 10 %, jatkohoito annetaan hoitavan lääkärin harkinnan mukaisesti.
Konsolidaatio-keskisuuri riski
- Kirjaudu tai rekisteröidy kommentoidaksesi
Konsolidaatio-keskisuuri riski
Yhteensä annetaan kolme konsolidaatiohoitoa jos allogeenista kantasolujensiirtoa ei tehdä.
Kantasolujensiirto, jäännöstauti, gemtutsumabi otsogamisiini (GO)
- Jos sopiva luovuttaja on olemassa alle 61-vuotiaalle potilaalle, allogeeninen siirto tehdään 1. konsolidaatiohoidon jälkeen jäännöstaudista riippumatta. Jos induktiohoidolla on saavutettu morfologinen remissio, ennen siirtoa annettava 1. konsolidaatiohoito on kevyempi kuin muiden potilaiden 1. konsolidaatiohoito. Hyväkuntoisilla yli 61-vuotiailla potilailla, joilla on tautinsa puolesta hyvin painava indikaatio allogeeniseen siirtoon, se voidaan tehdä kevennettyä esihoitoa käyttäen. Siirtoedellytykset harkitaan yksilöllisesti.
- Jos luovuttajaa ei ole eikä 1. konsolidaatiohoidon jälkeen todeta jäännöstautia, annetaan yhteensä kolme konsolidaatiohoitoa.
- Jos sisarus- eikä rekisterisiirto ole mahdollinen potilaalle, jolla 1. konsolidaatiohodon jälkeen on jäännöstauti, annetaan 2. ja 3. konsolidaatiohoidon yhteydessä GO-hoito.
Kevennetty 1. konsolidaatio allogeeniseen sisarussiirtoon meneville remissiopotilaille
- Sytarabiini 500 mg/m2 3 tunnin iv-infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Idarubisiini 8 mg/m2 15 min iv-infuusiona kerran päivässä päivinä 5 - 6.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
|
Lääke |
Annos |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Sytarabiini |
500 mg/m2 |
3 t inf |
x 2 |
1 – 4 |
|
Idarubisiini |
8 mg/m2 |
15 min inf |
x 1 |
5 – 6 |
|
Kortikosteroidisilmätipat |
1 gtt |
|
x 4 |
1 – 4 |
1. konsolidaatio muille kuin allogeeniseen sisarussiirtoon meneville remissiopotilaille, HDAraC+Ida
Alle 60-vuotiaat potilaat
- Sytarabiini 1500 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Idarubisiini 8 mg/m2 15 minuutin infuusiona kerran päivässä päivinä 5 – 7, yhteensä 3 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
|
Lääke |
Annos |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Sytarabiini |
1500 mg/m2 |
3 t inf |
x 2 |
1 – 4 |
|
Idarubisiini |
8 mg/m2 |
15 min inf |
x 1 |
5 – 7 |
|
Kortikosteroidisilmätipat |
1 gtt |
|
x 4 |
1 – 4 |
Jos 1. konsolidaatiohoidon jälkeen todetaan jäännöstauti, tehdään allogeeninen kantasolujensiirto myös rekisteriluovuttajalta mahdollisuuksien mukaan.
60-vuotta täyttäneet potilaat
- Sytarabiini 750 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Idarubisiini 8 mg/m2 kerran päivässä päivinä 5 – 6, yhteensä 2 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
|
Lääke |
Annos |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Sytarabiini |
750 mg/m2 |
3 t inf |
x 2 |
1 – 4 |
|
Idarubisiini |
8 mg/m2 |
15 min inf |
x 1 |
5 – 6 |
|
Kortikosteroidisilmätipat |
1 gtt |
|
x 4 |
1 – 4 |
2. konsolidaatio, Mito-HDAraC
Alle 60-vuotiaat potilaat
- Mitoksantroni 12 mg/m2 30 minuutin infuusiona, kerran päivässä päivinä 2 – 5, yhteensä 4 annosta.
- Sytarabiini 1500 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
- Jos 1. konsolidaatiohoidon jälkeen todettiin jäännöstauti eikä kantasolujensiirtoon ole mahdollisuutta, annetaan lisäksi gemtutsumabi otsogamisiini 5 mg (pinta-ala alle 1,7 m2) tai 10 mg (pinta-ala vähintään 1,7 m2) 2 tunnin infuusiona päivänä 1.
- Jos 1. konsolidaatiohoidon jälkeen todettiin jäännöstauti tehdään kantasolujensiirto 2. konsolidaation jälkeen mahdollisuuksien mukaan, jos sitä ei tehty 1. konsolidaation jälkeen.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Mitoksantroni |
12 mg/m2 |
30 min inf |
x 1 |
2 – 5 |
|
Sytarabiini |
1500 mg/m2 |
3 t inf |
x 2 |
1 – 4 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 – 4 |
|
Gemtutsumabi otsogamisiini osalle potilaista |
5 tai 10 mg/potilas ks yllä |
2 t inf |
1 |
1 |
60-vuotta täyttäneet potilaat
- Mitoksantroni 8 mg/m2 30 minuutin infuusiona kerran päivässä päivinä 2 – 5, yhteensä 4 annosta.
- Sytarabiini 750 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
- Jos 1. konsolidaatiohoidon jälkeen todettiin jäännöstauti, annetaan lisäksi gemtutsumabi otsogamisiini 5 mg (pinta-ala alle 1,7 m2) tai 10 mg (pinta-ala vähintään 1,7 m2) 2 tunnin infuusiona päivänä 1.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Mitoksantroni |
8 mg/m2 |
30 min inf |
x 1 |
2 – 5 |
|
Sytarabiini |
750 mg/m2 |
3 t inf |
x 2 |
1 – 4 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 – 4 |
|
Gemtutsumabi otsogamisiini osalle potilaista |
5 tai 10 mg/potilas ks yllä |
2 t inf |
1 |
1 |
3. konsolidaatio, MACE
- M-amsakriini 100 mg/m2 kerran päivässä 2 tunnin infuusiona päivinä 1 – 5 yhteensä 5 annosta.
- Sytarabiini 200 mg/m2/vrk jatkuvana infuusiona päivinä 1 - 5. (voidaan keskeyttää etoposidin ja amsakriinin annon ajaksi).
- Etoposidi (etoposidifosfaattina) 100 mg/m2 kerran päivässä 20 minuutin infuusiona päivinä 1 – 5, yhteensä 5 annosta.
Jos 1. konsolidaatiohoidon jälkeen todettiin jäännöstauti eikä kantasolujensiirtoon ole mahdollisuutta, annetaan lisäksi gemtutsumabi otsogamisiini 5 mg (pinta-ala alle 1,7 m2) tai 10 mg (pinta-ala vähintään 1,7 m2) infuusiona päivänä 1.
P-K oltava yli 3,7 ennen amsakriini-hoidon antamista.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Amsakriini |
100 mg/m2 |
2 t inf |
x 1 |
1 – 5 |
|
Sytarabiini |
200 mg/m2 |
24 t inf |
jatkuva inf |
1 – 5 |
|
Etoposidi |
100 mg/m2 |
20 min inf |
x 1 |
1 – 5 |
|
Gemtutsumabi otsogamisiini osalle potilaista |
5 tai 10 mg/potilas ks yllä |
2 t inf |
1 |
1 |
Neuroleukemian profylaksi
Neuroleukemian ennaltaehkäisevä hoito annetaan vain seuraaviin riskiryhmiin kuuluville potilaille:
- Kromosomin 16 inversio
- Akuutti myelomonosyyttileukemia tai monosyyttileukemia (FAB-tyyppi M4 tai M5) ja diagnoosivaiheen B-leuk > 40 x 109/l
- Akuutti myelomonosyyttileukemia tai monosyyttileukemia (FAB-tyyppi M4 tai M5) ja ekstramedullaarinen infiltraatti (mm. iho, imusolmukkeet yli 3 cm, perna).
Remissioon pääsyn jälkeen ja trombosyyttipitoisuuden ollessa yli 40 x 109/l annetaan sytarabiinia intratekaalisesti 100 mg yhteensä 5 annosta. It.-hoitojen väli on vähintään 1 viikko. Iv. ja it. annon välin tulee olla vähintään 48 tuntia, kun iv. infuusiona annettavan sytarabiinin annos on yli 500 mg/m2.
Konsolidaatio-pieni riski
- Kirjaudu tai rekisteröidy kommentoidaksesi
Konsolidaatio-pieni riski
Yhteensä annetaan 3 konsolidaatiohoitoa, joista 2. ja 3. konsolidaatiohoidon annokset ovat pienempiä kuin keskisuuren ja suuren riskin hoidoissa.
Jäännöstauti, kantasolujensiirto, gemtutsumabi otsogamisiini (GO)
Jos todetaan jäännöstauti ennen toista konsolidaatiohoitoa (yli 1 %) tai ennen 3. konsolidaatiohoitoa (yli 0,3 %), tehdään alle 61-vuotiaille potilaille allogeeninen kantasolujensiirto sisarukselta tai rekisteriluovuttajalta mahdollisuuksien mukaan. Hyväkuntoisille yli 61-vuotiaille potilaille, joilla on tautinsa puolesta hyvin painava indikaatio allogeeniseen siirtoon, voidaan se tehdä kevennettyä esihoitoa käyttäen. Siirtoedellytykset harkitaan yksilöllisesti.
Jos ennen 2 konsolidaatiohoitoa todetaan jäännöstauti (yli 0,3 %) eikä allogeeninen siirto ole mahdollinen, annetaan 2. ja 3. konsolidaatiohoito keskiriskin solunsalpaaja-annoksin ja niiden yhteydessä GO-hoito.
1. konsolidaatio HDAraC + Ida
Alle 60-vuotiaat potilaat
- Sytarabiini 1500 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Idarubisiini 8 mg/m2 15 minuutin infuusiona kerran päivässä päivinä 5 – 7, yhteensä 3 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
|
Lääke |
Annos |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Sytarabiini |
1500 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Idarubisiini |
8 mg/m2 |
15 min inf |
x 1 |
5 - 7 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
60-vuotta täyttäneet potilaat
- Sytarabiini 750 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Idarubisiini 8 mg/m2 15 minuutin infuusiona kerran päivässä päivinä 5 - 6.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Sytarabiini |
750 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Idarubisiini |
8 mg/m2 |
15 min inf |
x 1 |
5 - 6 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
2. konsolidaatio Mito-IDAraC
Ennen toista konsolidaatiohoitoa todetun jäännöstaudin vaikutus hoitoon: ks yllä!
Alle 60-vuotiaat potilaat
- Mitoksantroni 12 mg/m2 30 minuutin infuusiona kerran päivässä päivinä 2 - 5.
- Sytarabiini 1000 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1-4.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Mitoksantroni |
12 mg/m2 |
30 min inf |
x 1 |
2 - 5 |
|
Sytarabiini |
1000 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
60-vuotta täyttäneet potilaat
- Mitoksantroni 8 mg/m2 30 minuutin infuusiona kerran päivässä päivinä 2 - 5.
- Sytarabiini 750 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Mitoksantroni |
8 mg/m2 |
30 min inf |
x 1 |
2 - 5 |
|
Sytarabiini |
750 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
3. konsolidaatio MACE
Ennen toista konsolidaatiohoitoa todetun jäännöstaudin vaikutus hoitoon: ks yllä!
- M-amsakriini 100 mg/m2 kerran päivässä 2 tunnin infuusiona päivinä 1 - 4.
- Sytarabiini 100 mg/m2/vrk jatkuvana infuusiona päivinä 1 - 5 (voidaan keskeyttää etoposidin ja amsakriinin annon ajaksi).
- Etoposidi (etoposidifosfaattina) 100 mg/m2 kerran päivässä 20 minuutin infuusiona päivinä 1-5.
- P-K on oltava yli 3,7 ennen amsakriini-hoidon antamista.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Amsakriini |
100 mg/m2 |
2 t inf |
x 1 |
1 – 4 |
|
Sytarabiini |
100 mg/m2 |
jatkuva inf |
jatkuva inf |
1 – 5 |
|
Etoposidi |
100 mg/m2 |
20 min inf |
x 1 |
1 – 5 |
Neuroleukemian profylaksi
Neuroleukemian ennaltaehkäisevä hoito annetaan vain seuraaviin riskiryhmiin kuuluville potilaille:
- Kromosomin 16 inversio
- Akuutti myelomonosyyttileukemia tai monosyyttileukemia (FAB-tyyppi M4 tai M5) ja diagnoosivaiheen B-leuk > 40 x 109/l
- Akuutti myelomonosyyttileukemia tai monosyyttileukemia (FAB-tyyppi M4 tai M5) ja ekstramedullaarinen infiltraatti (mm. iho, imusolmukkeet yli 3 cm, perna).
Remissioon pääsyn jälkeen ja trombosyyttipitoisuuden ollessa yli 40 x 109/l annetaan sytarabiinia intratekaalisesti 100 mg yhteensä 5 annosta. It.-hoitojen väli on vähintään 1 viikko. Iv. ja it. annon välin tulee olla vähintään 48 tuntia, kun iv. infuusiona annettavan sytarabiinin annos on yli 500 mg/m2.
Konsolidaatio-suuri riski
- Kirjaudu tai rekisteröidy kommentoidaksesi
Konsolidaatio-suuri riski
Jäännöstauti, kantasolujensiirto, gemtutsumabiotsogamisiini (GO)
Denovo tai sekundaari leukemia
Jos alle 61-vuotiaalle potilaalle on olemassa luovuttaja, allogeeniseen siirtoon mennään 1. konsolidaatiohoidon jälkeen. Hyväkuntoisilla yli 61-vuotiailla potilailla, joilla on tautinsa puolesta hyvin painava indikaatio allogeeniseen siirtoon, se voidaan tehdä kevennettyä esihoitoa käyttäen. Siirtoedellytykset harkitan yksilöllisesti.
Jos allogeeninen siirto ei ole mahdollinen, potilas saa 4 konsolidaatiohoitoa.
Jos jäännöstauti todetaan 1.konsolidaatiohoidon jälkeen otetussa näytteessä, annetaan 2.- 4. konsolidaatiohoidon yhteydessä gemtutsumabi otsogamisiini -hoito, jos kantasolujensiirtoon ei ole mahdollisuutta.
MDSAML
Allogeeninen siirto tehdään heti kun sen on mahdollista ja blastien osuus on korkeintaan 10 %. Tarvittaessa annetaan ennen siirtoa redusoitu konsolidaatiohoito.
Jos allogeeninen siirto ei ole mahdollinen, annetaan tarpeen mukaan redusoidut konsolidaatiohoidot ohjelman mukaisesti.
1. konsolidaatio, HDAraC-Ida
Alle 60-vuotiaat potilaat
- Sytarabiini 1500 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 – 4, yhteensä 8 annosta.
- Idarubisiini 8 mg/m2 kerran päivässä 15 minuutin infuusiona päivinä 5 – 7, yhteensä 3 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
|
Lääke |
Annos |
Antoaika ja -tapa |
Antokerrat/pv |
Antopäivät |
|
Sytarabiini |
1500 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Idarubisiini |
8 mg/m2 |
15 min inf |
x 1 |
5 - 7 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
Kantasolujensiirto tehdään sisarusluovuttajalta 1. konsolidaatiohoidon jälkeen ja rekisteriluovuttajalta 1. tai 2. konsolidaatiohoidon jälkeen.
60-vuotta täyttäneet potilaat
- Sytarabiini 750 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1-4, yhteensä 8 annosta.
- Idarubisiini 8 mg/m2 kerran päivässä 15 minuutin infuusiona päivinä 5-6, yhteensä 2 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1-4.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Sytarabiini |
750 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Idarubisiini |
8 mg/m2 |
15 min inf |
x 1 |
5 - 6 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
2. konsolidaatio, Mito-HDAraC
Alle 60-vuotiaat potilaat
- Mitoksantroni 12 mg/m2 30 minuutin infuusiona kerran päivässä päivinä 2 - 5.
- Sytarabiini 1500 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
- Jos 1. konsolidaatiohoidon jälkeen todettiin jäännöstauti, annetaan lisäksi gemtutsumabi otsogamisiinia 5 mg (pinta-ala alle 1,7 m2) tai 10 mg (pinta-ala vähintään 1,7 m2) infuusiona päivänä 1, jos allogeeniseen kantasolujensiirtoon ei ole mahdollisuutta.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Mitoksantroni |
12 mg/m2 |
30 min inf |
x 1 |
2 - 5 |
|
Sytarabiini |
1500 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
|
Gemtutsumabi otsogamisiini osalle potilaista |
5 tai 10 mg/potilas ks yllä |
2 t inf |
1 |
1 |
60-vuotta täyttäneet potilaat
- Mitoksantroni 8 mg/m2 30 minuutin infuusiona kerran päivässä päivinä 2 - 5.
- Sytarabiini 750 mg/m2 3 tunnin infuusiona 12 tunnin välein päivinä 1 - 4, yhteensä 8 annosta.
- Kortikosteroidisilmätipat 1gtt x 4/vrk kumpaankin silmään päivinä 1 - 4.
- Jos 1. konsolidaatiohoidon jälkeen todettiin jäännöstauti, annetaan lisäksi gemtutsumabi otsogamisiinia 5 mg (pinta-ala alle 1,7 m2) tai 10 mg (pinta-ala vähintään 1,7 m2) 2 tunnin infuusiona päivänä 1.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Mitoksantroni |
8 mg/m2 |
30 min inf |
x 1 |
2 - 5 |
|
Sytarabiini |
750 mg/m2 |
3 t inf |
x 2 |
1 - 4 |
|
Kortikosteroi-disilmätipat |
1 gtt |
|
x 4 |
1 - 4 |
|
Gemtutsumabi otsogamisiini osalle potilaista |
5 tai 10 mg/potilas ks yllä |
2 t inf |
1 |
1 |
3. konsolidaatio, MACE
- M-amsakriini 100 mg/m2 kerran päivässä 2 tunnin infuusiona päivinä 1-5.
- Sytarabiini 200 mg/m2 /vrk jatkuvana infuusiona päivinä 1-5 (voidaan keskeyttää etoposidin ja amsakriinin annon ajaksi).
- Etoposidi (etoposidifosfaattina) 100 mg/m2 kerran päivässä 20 minuutin infuusiona päivinä 1-5.
Jos 1. konsolidaatiohoidon jälkeen todettiin jäännöstauti, annetaan lisäksi gemtutsumabi otsogamisiinia 5 mg (pinta-ala alle 1,7 m2) tai 10 mg (pinta-ala vähintään 1,7 m2) 2 tunnin infuusiona päivänä 1, jos allogeeniseen kantasolujensiirtoon ei ole mahdollisuutta.
P-K on oltava yli 3,7 ennen amsakriini-hoidon antamista.
|
Lääke |
Annos |
Antoaika ja –tapa |
Antokerrat/pv |
Antopäivät |
|
Amsakriini |
100 mg/m2 |
2 t inf |
x 1 |
1 - 5 |
|
Sytarabiini |
200 mg/m2 |
jatkuva inf |
jatkuva inf |
1 – 5 |
|
Etoposidi |
100 mg/m2 |
20 min inf |
x 1 |
1 – 5 |
|
Gemtutsumabi otsogamisiini osalle potilaista |
5 tai 10 mg/potilas ks yllä |
2 t inf |
1 |
1 |
4. konsolidaatioICE
- Idarubisiini 8 mg/m2 15 minuutin infuusiona päivinä 2 - 4, yhteensä 3 annosta.
- Sytarabiini 200 mg/m2 jatkuvana infuusiona päivinä 1 - 5. Infuusio voidaan keskeyttää etoposidin ja idarubisiinin annon ajaksi.
- Etoposidi (etoposidifosfaattina) 100 mg/m2 20 minuutin infuusiona päivinä 1 – 5, yhteensä 5 annosta.
Jos 1. konsolidaatiohoidon jälkeen todettiin jäännöstauti, annetaan lisäksi gemtutsumabi otsogamisiinia 5 mg (pinta-ala alle 1,7 m2) tai 10 mg (pinta-ala vähintään 1,7 m2) 2 tunnin infuusiona päivänä 1, jos allogeeniseen kantasolujensiirtoon ei ole mahdollisuutta.
|
Lääke |
Annos |
Antoaika |
Antokerrat/pv |
Antopäivät |
|
Idarubisiini |
8 mg/m2 |
15 min inf |
x 1 |
2 – 4 |
|
Sytarabiini |
200 mg/m2 |
24 t inf |
jatkuva inf |
1 - 5 |
|
Etoposidi |
100 mg/m2 |
20 min inf |
x 1 |
1 - 5 |
|
Gemtutsumabi otsogamisiini osalle potilaista |
5 tai 10 mg/potilas ks yllä |
2 t |
x 1 |
1 |
Neuroleukemian profylaksi
Neuroleukemian ennaltaehkäisevä hoito annetaan vain seuraaviin riskiryhmiin kuuluville potilaille:
- Kromosomi 16 inversio
- Akuutti myelomonosyyttileukemia tai monosyyttileukemia (FAB-tyyppi M4 tai M5) ja diagnoosivaiheen B-leuk > 40 x 109/l
- Akuutti myelomonosyyttileukemia tai monosyyttileukemia (FAB-tyyppi M4 tai M5) ja ekstramedullaarinen infiltraatti (mm. iho, imusolmukkeet yli 3 cm, perna).
Remissioon pääsyn jälkeen ja trombosyyttipitoisuuden ollessa yli 40 x 109/l annetaan sytarabiinia intratekaalisesti 100 mg yhteensä 5 annosta. It.-hoitojen väli on vähintään 1 viikko. Iv. ja it. annon välin tulee olla vähintään 48 tuntia, kun iv. infuusiona annettavan sytarabiinin annos on yli 500 mg/m2.
Riskiluokitus
- Kirjaudu tai rekisteröidy kommentoidaksesi
Riskiluokitus
Potilaat jaetaan 1) pienen, 2) keskisuuren ja 3) suuren riskin ryhmiin diagnoosivaiheen löydösten ja ensimmäisen induktiohoidon tuloksen perusteella.
Pieni riski
Pienen riskin ryhmään kuuluvat ne potilaat, joiden kohdalla kaikki alla olevat kriteerit täyttyvät.
- De novo tai sekundaarileukemia
- Karyotyyppi
- t(8;21), inv(16)(p13;q22) tai t(16;16)(p13;q22) ja
- korkeintaan yksi muu poikkeavuus, joka ei saa olla suuren riskin poikkeavuus.
- Muut kriteerit
- Diagnoosivaiheen leukosyyttien pitoisuus on alle 100 x 109/l.
- Diagnoosivaiheessa ei ole ekstramedullaarista leukemiaa.
- Ensimmäisen induktiohoidon jälkeen luuytimen blastien osuus on korkeintaan 10 %.
Keskisuuri riski
Keskisuuren riskin ryhmään kuuluvat ne potilaat, joiden kohdalla kaikki alla olevat kriteerit täyttyvät.
- De novo leukemia
- Karyotyyppi
- normaali tai ei määritettävissä
- +8, -Y, +6, del(12p) tai muu, mutta suureen riskiin kuulumaton poikkeavuus.
- poikkeavuutta lukuun ottamatta pienen ja suuren riskin ryhmään kuuluvia poikkeavuuksia.
- Muut kriteerit
- Diagnoosivaiheessa leukosyyttien pitoisuus on alle 100 x 109/l
- Diagnoosivaihessa ei ole ekstramedullaarisia infiltraatioita (esimerkiksi iholeukemia, keskushermostoleukemia, yli 3 cm läpimittaisia imusolmukkeita, pernan pisin mitta yli 16 cm, useita kloroomia).
- Esimmäisen induktiohoidon jälkeen luuytimen blastien osuus on korkeintaan 10 %.
Suuri riski
De novo tai sekundaarileukemia. Suuren riskin ryhmään kuuluvat ne potilaat, joiden kohdalla yksikin alla olevista kriteereistä täyttyy.
- Karyotyyppi
- 5, del(5q), -7, del(7q), inv(3q), t(3q21-26), poikkeava 11q23, 20q, 21q, 17p, del(9q), t(6;9), t(9;22).
- 3 poikkeavuutta.
- Jokin edellä mainittu suuren riskin poikkeavuus ja lisäksi jokin pienen tai keskisuuren riskin poikkeavuus.
- Sekundaarileukemia, jossa on keskisuuren riskin karyotyyppi, karyotyyppi on normaali tai karyotyyppiä ei ole määritetty.
- Muut kriteerit
- B-leuk diagnoosivaiheessa yli 100 x 109/l.
- Ekstramedullaarinen leukemian manifestaatio.
- Minimaalisesti erilaistunut AML (FAB M0), akuutti erytroleukemia (FAB M6), ja akuutti megakaryosyyttileukemia (FAB M7) sekä akuutti hybridileukemia modifioidun EGIL-luokituksen mukaan.
- Monilinjainen dysplasia WHO:n luokituksen mukaan.
- Ensimmäisen induktiohoidon jälkeen luuytimen blastien osuus on yli 10 %.
- Myelodysplastisen oireyhtymän tai myelodysplastisen myeloproliferatiivisen taudin jälkeen kehittynyt AML. Edellytyksenä on, että perustauti on ollut diagnosoituna 3 kk ennen AML-diagnoosin tekoa.
Epäselvä riski
Jos riskiluokitusta ei voida tehdä tietojen puuttuessa, potilaan katsotaan hoitovalintojen osalta kuuluvan keskisuuren riskin ryhmään.
Otsikko vaihdettiin muodosta <em class="placeholder">Riskiluokitus</em> muotoon <em class="placeholder">Riskiluokitus</em>.
Clo/Cyt/Eto
- Kirjaudu tai rekisteröidy kommentoidaksesi
Clo/Cyt/Eto
Käyttöaiheet
- relapsoitunut tai refraktaari AML tai ALL
Lääkkeet ja annokset
- Klofarabiini 40 mg/m2 iv pv 1-5
- Sytarabiini 1000 mg/m2 iv pv 1-5
- Etoposidi 100 mg/m2 iv pv 1-4
Infuusio-ohjeet
- Jos syklofosfamidi (tai sytarabiini) annostellaan 2 kertaa päivässä:
- klo 9 syklofosfamidi (1 tunnin infuusio) tai sytarabiini (1-3 tunnin infuusio)
- klo 13 etoposidi (15-20 minuutin infuusio)
- klo 16.30 hydrokortisoni 250 mg iv bolus
- klo 17 klofarabiini (1-2 tunnin infuusio)
- klo 21 syklofosfamidi (1 tunnin infuusio) tai sytarabiini (1-3 tunnin infuusio)
- Jos sytarabiini annostellaan kerran päivässä, sytarabiinin infuusio aloitetaan 4 tuntia klofarabiini-infuusion alusta
- klo 10 etoposidi (15-20 min. infuusio)
- klo 11.30 hydrokortisoni 250 mg iv bolus
- klo 12 klofarabiini (1-2 tunnin infuusio)
- klo 16 sytarabiini (1-3 tunnin infuusio)
Muistettavaa
Kirjallisuutta
- Report of a Phase II Study of Clofarabine and Cytarabine in De Novo and Relapsed and Refractory AML Patients and in Selected Elderly Patients at High Risk for Anthracycline Toxicity. Agura E, Cooper B, Holmes H, Vance E, Berryman RB, Maisel C, Li S, Saracino G, Tadic-Ovcina M, Fay J. Oncologist. 2011;16(2):197-206.
| Kirjoittajat: | Kimmo Porkka, Erkki Elonen, Anna Kosola |
FLAG-Ida
- Kirjaudu tai rekisteröidy kommentoidaksesi
FLAG-Ida
Indikaatio
Akuutit leukemiat, relapsin hoito.
Annostelu
|
Lääke |
Annos |
Antotapa |
Antokerrat/pv |
Antopäivät |
Annoksia yhteensä |
|
Idarubisiini |
10 mg/m2, 7 mg/m2 56 v täyttäneille |
15 min inf |
x 1 |
1 ja 3 |
2 |
|
Fludarabiini |
30 mg/m2 |
1 t infusiona 4 t ennen AraC |
x 1 |
1-5 |
5 |
|
Sytarabiini (AraC) |
2000 mg/m2 1000 mg/m2 56 v täyttäneille |
2 tunnin infuusiona 4 t fludarainfuusion lopettamisen jälkeen |
x 1 |
1-5 |
5 |
|
Filgrastiimi |
300 mg/potilas |
sc |
x 1 |
7 ® |
tarpeen mukaan |
Muitakin annosteluja käytetään
Kirjallisuutta
Kompetenznetz 17.1.2004
ICE
- Kirjaudu tai rekisteröidy kommentoidaksesi
ICE
Indikaatiot
- Akuutti myelooinen leukemia. relapsin hoito.
- Krooninen myelooinen leukemia, myelooinen blastikriisi.
Annostelu
- Idarubisiini 8 mg/m2 päivinä 1-5
- Sytarabiini 800 mg/m2 2 tunnin infuusiona päivinä 1-5
- Etoposidi 150 mg/m2 2 tunnin infuusiona päivinä 1-5
- Muitakin annosteluohjeita on samannimiselle hoidolle.
- KML-potilaista osa jää pitkäaikaisesti trombosytopeenisiksi
Kirjallisuutta
Blood 1995; 86 (10) suppl 1: #1615 ASH95 ja #3955 ASH95
Iäkkäiden AML
- Kirjaudu tai rekisteröidy kommentoidaksesi
Iäkkäiden AML
- Useimpia 66 vuotta täyttäneitä AML-potilaita ei voida hoitaa nuorempien hoito-ohjelmien mukaisin täysin annoksin. Monia voidaan kuitenkin hoitaa remissioon pyrkien. Näille potilaille annetaan AML2003 ohjelman hoitoja soveltaen. Granulosyyttikasvutekijän avulla neutropenia-aikaa voidaan lyhentää.
- Iäkkäiden (66 vuotta täyttäneiden) IAT
- IAT induktiohoito voidaan harkinnan mukaan antaa 5-7 päivän mittaisena ja idarubisiiniannoksia voidaan antaa 1-3.
Idarubisiini
- 12 mg/m2 yhtenä, kahtena tai kolmena päivänä (päivät 1, 3 ja 5) 15 minuutin infuusiona, yhteensä 1-3 annosta.
Sytarabiini
- 50 mg/m2 15 min infuusiona päivänä 1 sekä jatkuvana infuusiona 100 mg/m2/vrk alkaen päivänä 1, harkinnan mukaan 5-7 vrk:n ajan.
Tioguaniini
- 75 mg/m2 suun kautta 12 tunnin välein alkaen päivänä 1, harkinnan mukaan 5-7 päivänä, yhteensä 10-14 annosta.
- Jatkohoito riippuu hoitovasteesta ja hoidon siedosta. Usein hoitoa voidaan jatkaa AML2003 konsolidaatiohoidoin modifioiden ja redusoiden.
- Iäkkäiden (66 vuotta täyttäneiden) MEA
- Iäkkäille potilaille soveltuu usein 56 vuotta täyttäneiden MEA-hoito 75-prosenttisin annoksin siten että kutakin lääkettä annetaan kolmena päivänä neljän päivän sijasta.
Mitoksantroni
- 8 mg/m2 30 minuutin inf. päivinä 3 ‑ 5.
Etoposidifosfaatti, annos vastaa etoposidia
- 100 mg/m2 15 - 20 minuutin inf. päivinä 1 ‑ 3. Jos annetaan etoposidia, infuusioaika on 1 tunti.
Sytarabiini
- 400-500 mg/m2 2 tunnin infuusiona 12 tunnin välein päivinä 1 ‑ 3, yhteensä 6 annosta
Jatkohoito riippuu hoitovasteesta ja hoidon siedosta. Usein hoitoa voidaan jatkaa AML2003 konsolidaatiohoidoin modifioiden ja redusoiden.
Kirjallisuutta
Kirjallisuutta
Hoitosuosituksia
- Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Löwenberg B, Bloomfield CD; European LeukemiaNet. Blood. 2010 Jan 21;115(3):453-74
Harvinaiset lymfoproliferatiiviset taudit
Harvinaiset lymfoproliferatiiviset taudit
EBV-lymfoproliferaatio
- Kirjaudu tai rekisteröidy kommentoidaksesi
EBV-lymfoproliferaatio
Luokitus: ICD-O xxx, ICD-10 xxx
Ebstein-Barr-viruksen (EBV) aiheuttama lymfoproliferatiivinen tauti liittyy yleisimmin kiinteiden elinten siirron tai hematopoieettisen kantasolujensiirron jälkeiseen soluvälitteisen immuunivasteen häiriöön, johtuen pääosin käytetystä immunosuppressiivisesta lääkityksestä. Tilasta käytetään nimeä elinsiirron jälkeinen lymfoproliferatiivinen tauti (post-transplantation lymphoproliferative disease, PTLD).
(lyhyesti, yhteenvetomaisesti taustaa, diagnostiikkaa ja hoitoa)
| Kirjoittajat: | Mervi Saari, Kimmo Porkka |
| © Suomen Hematologiyhdistys | Suomen Leukemiaryhmä |
Diagnoosi
- Kirjaudu tai rekisteröidy kommentoidaksesi
Diagnoosi
Epäily PTLD-taudista herää ... vuoksi.
Diagnoosi perustuu oireisiin, ....
Oireet ja löydökset
- kuume
Verikokeet
- TVK, CRP, La, ALAT, ASAT, Afos, LD, Krea
- P-RF, S-ANAAb, E-Coomb-O, S-Prot-Fr, S-CMVAb
Veren ja luuytimen tutkimukset
- luuydinaspiraatti, josta
- luuydinbiopsian histologinen tutkimus ja immunohistokemiallinen tutkimus lymfosyytti-infiltraatioasteen määrittämiseksi
Kuvantamistutkimukset
- vartalon TT
- thorax
Erotusdiagnoosi
- ...
Hoito
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hoito
Useimmat potilaat tarvitsevat hoitoa jossain sairautensa vaiheessa. Hoidon aiheita ovat ...
Ensilinjan hoito
- Immunosuppression purkaminen...
- Rituksimabi (annos, kuinka monta kertaa)
Hoidon vaste tulee yleensä hitaasti, kuukausien kuluessa
Hoidon vaihtamisen aiheita:
- Ei vastetta 3(-6) kk hoidolle
- Lääkitykseen liittyvät sivuoireet
Toisen ja myöhemmän linjan hoito
- Tekstiä
Hoitokaavio
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hoitokaavio
PTLD:n hoitokaavio (Heslop HE. Blood. 2009 114:4002-8):
LGL-leukemia
LGL-leukemia
| Liite | Koko |
|---|---|
| 980.87 KB |
Autoimmuunisytopeniat
Autoimmuunisytopeniat
Anemiat
Anemiat
AIHA
AIHA
Autoimmuunihemolyyttinen anemia
Hemolyysin diagnoosi
Hemolyysin diagnoosi
Oireet
Anemiaoireet, kellertävä iho. Tumma virtsa intravaskulaarisessa hemolyysissa. Selkä-ja vatsakivut (väärän) verensiirron yhteydessä. Tromboositaipumus PNH:ssa ja sirppisoluanemiassa. Mikroangiopaattisissa hemolyyttisissa anemioissa: trombosytopenia, hematuria, munuaisten vajaatoiminta (HUS/TTP) keskushermosto-oireet (TTP), kärkijäsenten syanoosi-nekroosi (DIC)
Laboratoriodiagnostiikka
Hb matala, usein lievää makrosytoosia (retikulosyyteistä)
Retikulosyytit koholla (jos luuydin kunnossa)
LD koholla
S-bilirubiini (konjugoimaton) koholla
S-haptoglobiini laskee (huom! akuutin faasin proteiinina saattaa olla koholla)
E-Coombs positiivinen AIHA:ssa, osoittaa punasolujen pinnalle kiinnittyneen vasta-aineen ja/tai komplementin
Veren sivelyvalmiste, B-morfo: fragmentaatiota mikroangiopaattisissa tiloissa, sferosyyttejä synnynnäisessä sferosytoosissa ja AIHA:ssa
Immunoglobuliinimääritykset (S-IgGAM): joskus AIHA ensimmäinen merkki CVID-taudista (common variable immunodeficiency)
Hoito
Hoito
Hemolyyttisen kriisin akuuttihoito
Vaikean anemian hoitona on punasolujen siirto välittömästi. Nopea Hb:n lasku voi johtaa instabiiliin hemodynamiikkaan tai henkeä uhkaavaan hypoksemiaan. Punasoluja annettaessa tulee varoa verenkierron ylikuormittumista ja keuhkoödeemaa.
Annetaan parhaiten sopivia ABO- ja Rh-ryhmän mukaisia Kell-negat punasoluja postitiivisesta sopivuuskokeesta huolimatta. Immunohemolyysissa voi olla vaikeaa löytää sopivaa verta solujen pinnassa olevan vasta-aineen takia.
Kortikosteroidisuoja, esim. hydrokortisoni 100 mg iv. Punasolujen anto lopetetaan, kun kriittiset oireet helpottuvat.
Tiheä Hb:n seuranta (esim. 4-6 t välein)
Riiittävä hapetus ja nesteytys
Lämmin-AIHA
Peruslääke on kortikosteroidi: metyyyliprednisoloni (tai vastaava annos muuta) 1-2 mg/kg/vrk Vaikeassa tapuksessa aloitetaan iv, jatkossa po. Vaste pitäisi näkyä muutamassa päivässä.
Hätätilanteessa plasmanvaihto.
Huonosti reagoivat tapaukset:
Gammaglobuliini 0.4 mg/ kg iv 5 vrk
Rituksimabi 375mg/m2 iv kerran viikossa 4 viikon ajan
Jatkohoidossa: siklosporiini, atsatiopriini, syklofosfamidi, danatsoli tai pernan poisto
Kylmä-AIHA
Potilas pidettävä lämpimänä ja verituotteet ja infuusioliuokset lämmitettävä.
Kortisoni, iv-gammaglobuliini sekä pernan poisto tehoavat huonosti. Rituksimabi 375mg/m2 kerran viikossa 4 viikon ajan tehoaa osalla.
Jos liittyy esim. mykoplasma-tai virusinfektioon, korjaantuu infektion myötä.
Jos liittyy lymfoproliferatiivisiin sairauksiin, solunsalpaaja saattaa korjata.
Vaikeassa plasmanvaihto.
Aplastinen anemia
Aplastinen anemia
| Liite | Koko |
|---|---|
| 377.03 KB |
ITP
ITP
Immunologinen (idiopaattinen) trombosytopenia
Immunologisessa trombosytopeniassa (ITP) potilaan immuunijärjestelmä muodostaa autovasta-aineita trombosyyttien ja ilmeisesti myös luuytimen megakaryosyyttien pinta-antigeeneja kohtaan.
Vasta-aineilla päällystetyt trombosyytit fagosytoidaan lähinnä pernassa, ja perifeerisessä veressä todetaan trombosytopenia. Myös T-soluvälitteinen immuniteetti on häiriintynyt.
Trombosyyttien elinikä on lyhentynyt ja on usein alle 1 vrk. Oirekuva vaihtelee oireettomuudesta tai ainoastaan petekkia- ja mustelmataipumuksesta, joskus vakaviinkin vuotoihin. ITP-potilaat sietävät pieniäkin trombosyyttilukemia huomattavasti paremmin kuin luuydininfiltraation tai –aplasian aiheuttamaa trombosytopeniaa sairastavat. Trombosyyttitasolla ≥ 10 x 109/l esiintyy hyvin harvoin vakavia vuotoja. Jos trombosyytit ovat > 100 x 109/l, ei tutkimuksiin yleensä ole aihetta ryhtyä. Mitään spesifistä diagnostista testiä ITP:lle ei ole, vaan se on poissulkudiagnoosi.
Alla liitteenä SHY:n Infektiot ja immuniteetti- ryhmän päivitetty hoitosuositus.
| Liite | Koko |
|---|---|
| 392.72 KB |
KLL
KLL
Luokitus: KLL ICD-O 9823/3, ICD-10 C91.1 B-SLL ICD-O 9670/3, ICD-10 C83.00
 Krooninen lymfaattinen leukemia (KLL) on pahanlaatuinen veritauti, jolle on ominaista morfologisesti kypsien, mutta toiminnaltaan vajavaisten klonaalisten B-lymfosyyttien kertyminen elimistöön: luuytimeen, perifeeriseen vereen, imusolmukkeisiin ja pernaan. Termiä pienilymfosyyttinen lymfooma (SLL) käytetään WHO-luokituksen mukaan niistä KLL-tapauksista, joissa kasvaimen morfologia ja immunofenotyyppi vastaa KLL:aa, mutta perifeerisessä veressä ei todeta lymfosytoosia eikä potilaalla ole sytopenioita.
Krooninen lymfaattinen leukemia (KLL) on pahanlaatuinen veritauti, jolle on ominaista morfologisesti kypsien, mutta toiminnaltaan vajavaisten klonaalisten B-lymfosyyttien kertyminen elimistöön: luuytimeen, perifeeriseen vereen, imusolmukkeisiin ja pernaan. Termiä pienilymfosyyttinen lymfooma (SLL) käytetään WHO-luokituksen mukaan niistä KLL-tapauksista, joissa kasvaimen morfologia ja immunofenotyyppi vastaa KLL:aa, mutta perifeerisessä veressä ei todeta lymfosytoosia eikä potilaalla ole sytopenioita.
KLL on länsimaiden yleisin leukemia ja sen osuus kaikista leukemioista on vajaa kolmannes. Suomessa diagnosoidaan vuosittain n. 150 uutta tapausta. Sairastuneiden mediaani-ikä on 72 vuotta. KLL on heterogeeninen sairaus, jonka kulku vaihtelee indolentista hyvin aggressiiviseen. Sairauden kliinisella vaiheella ja sytogeneettisilla muutoksilla on merkittävä vaikutus ennusteeseen.
KLL todetaan usein sattumalöydöksenä, kun verenkuvassa on todettu leukosytoosi ja lymfosytoosi. Potilas voi hakeutua tutkimuksiin myös imusolmukesuurentumien vuoksi. Yleisoireet kuten väsymys, laihtuminen ja lämpöily liittyvät yleensä pitkälle edenneeseen tautiin. KLL potilailla esiintyy tavanomaista enemmän infektioita.
KLL voidaan diagnosoida perifeerisestä verestä. Immunofenotyypitys on keskeinen osa diagnostiikkaa. Diagnoosivaiheessa on tärkeää erottaa KLL muista lymfoproliferatiivisista sairauksista, kuten prolymfosyyttileukemiasta, lymfoplasmasyyttilymfoomasta, karvasoluleukemiasta, manttelisolulymfoomasta, pernan marginaalivyöhykkseen lymfoomasta, follikulaarisesta lymfoomasta ja LGL-leukemiasta.
| Kirjoittajat: | KLL- ryhmä |
| © Suomen Hematologiyhdistys | Suomen Leukemiaryhmä |
Diagnoosi
Diagnoosi
KLL-diagnoosiin vaaditaan krooninen monoklonaalinen B-lymfosytoosi: Perifeerisessä veressä klonaalisia KLL fenotyypin omaavia B-lymfosyyttejä ≥ 5,0 x 109/L (Poikkeus: ks. SLL). Veren B-lymfosyyttien klonaalisuus tulee varmentaa virtaussytometrialla. Jos lymfosyyteistä >55% on atyyppisiä prolymfosyyttejä, kyseessä on prolymfosyyttileukemia (ks taulukko 1).
Lyhytaikaista (<3kk) tai vähäistä (<5-10 x 10E9/l) lymfosytoosia ei tarvitse tutkia.
Luokittelu
Monoklonaalinen B-lymfosytoosi ( MBL)
Termiä monoklonaalinen B-lymfosyytosi (MBL) käytetään, jos perifeerisessä veressä todetaan klonaalisia, KLL-immunofenotyypin omaavia B-lymfosyyttejä vähemmän kuin 5,0 x 109/L eikä oireettomalla potilaalla ole lymfadenopatiaa, organomegaliaa tai sytopenioita. Monoklonaalisen kloonin tulee olla stabiili 3 kk ajan. MBL voidaan virtaussytometrisesti todeta n. 3-4% väestöstä ja 13-18%:lla KLL-potilaiden 1. asteen sukulaisista.
Jos klonaalisia B-lymfosyyttejä on 0.5 - 4.9 x 109/l, etenemisriski hoitoa vaativaksi KLL:ksi on 1-2% / vuosi.
Jos klonaalisia B-lymfosyyttejä on alle 0.5 x 109/l, tila etenee harvoin KLL:ksi.
Pienilymfosyyttinen lymfooma ( SLL)
Termiä pienilymfosyyttinen lymfooma (SLL) käytetään WHO-luokituksen mukaan niistä KLL-tapauksista, joissa kasvaimen morfologia ja immunofenotyyppi vastaa KLL:aa, mutta perifeerisessä veressä ei todeta lymfosytoosia eikä potilaalla ole sytopenioita. Mikäli potilaalla on luuydininfiltraatiosta johtuvia sytopenioita, dg:ksi voidaan asettaa KLL pienemmillä lymfosyyttimäärillä kuin 5,0 x 109/L.
Familiaalinen KLL
Noin 5%:lla KLL-potilaista on yksi tai useampia sukulaisia, joilla on KLL tai jokin muu lymfoproliferatiivinen sairaus. Silloin KLL luokitellaan familiaariseksi. Se ei eroa sporadisesta taudista kliinisesti tai ennusteen suhteen. KLL potilaiden ensimmäisen asteen sukulaisilla on n. 7-kertainen suhteellinen riski sairastua elämänsä aikana KLL:aan. Absoluuttisesti riski on kuitenkin vain 0.45 %
Diagnoosivaiheen tutkimukset
Anamneesi:
- Yleisoireet: kuumeilu, laihtuminen, yöhikoilu
- Suorituskyky
- Infektiot
- Tuberkuloosialtistus
- Vuoto-oireet
- Sukuanamneesi: lymfoproliferatiiviset sairaudet
Kliininen tutkimus:
- maksan ja pernan koko
- imusolmukestatus
Laboratoriotutkimukset
- B-PVKT+TKD
- B-morfo
- Veren lymfosyyttien immunofenotyypitys
Immunofenotyypitys
KLL:ssä B-lymfosyyteillä on tyypillinen immunofenotyyppi: CD19+, CD20+, CD23+ ja CD5+. Lisäksi B-solut ilmentävät heikosti pintaimmunoglobuliinia (joko kappa tai lambda). Tyypillistä on myös, että CD20 ja CD79b ilmentyvät heikosti ja että CD200 on positiivinen.
Taulukko 1. Lymfosyyyttien solumorfologian ja immunofenotyypin merkitys
(Frater JL, McCarron KF, Hammel JP, Shapiro JL, Miller ML, Tubbs RR, Pettay J, His E. Am J Clin Pathol 2001;116:655-6641)
|
Tauti |
Prolymfos. (%) |
Pieniä ja |
Atyyppisiä lymfos. |
Tyypillinen immunofenotyyppi |
|
KLL |
≤10 |
Ei |
<10 |
CD5+, CD19+, CD20+dim, |
|
KLL/PL
|
>10 ja <55 |
Ei |
- |
Kuten KLL |
|
PL |
≥55 |
Ei |
- |
CD5-/+,CD19+, CD20+, |
|
KLL, sekasoluinen |
≤10 |
Kyllä |
<10 |
Kuten KLL |
|
atyyppinen KLL
|
≤10 |
Ei |
≥10 |
Kuten KLL |
Solumorfologia, joka liittyy prolymfosyyttien tai atyyppisten lymfosyyttien lisääntyneeseen osuuteen, assosioituu sytogeneettisiin poikkeavuuksiin, useimmin trisomia 12 ja del17p ja epäsuotuisaan mutaatiostatukseen (unmutated IgVH status) (Swerlow SH, Campo E, Harris NL, Jaffe ES, Pileci SA, Stein H, Thiele J, Vardiman JW, eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th edn. Lyon, France: International Agency for Research on Cancer (IARC); 2008.) ja progressiiviseen tautiin ja terapialle refraktaarisuuteen (Marionneaux S et al 2014) .
Taulukko 2. CD5 –positiivisten lymfoproliferatiivisten tautien immunofenotyypityksen eroja
|
|
CLL |
MCL |
SMZL |
B PLL |
LPL |
|
CD20 (MFI) |
+ dim |
moderate/ bright |
moderate/ bright |
moderate/ bright |
moderate |
|
CD5 (%) |
80 |
90 |
20 |
30 |
15 |
|
CD23 (%) |
90 |
10 |
20 |
15 |
20 |
|
sIg (MFI) |
dim |
bright |
bright |
bright |
variable |
|
CD79b |
neg/dim |
pos |
pos |
pos |
pos |
MFI = mean fluorescence intensity
% viittaa antigeeniä ilmentävien solujen osuuteen tautitapauksissa
ref. Practical Flow Cytometry in Haematology Diagnosis. First Edition. M. Leach, M. Drummond and A. Doig (2013), Wiley-Blackwell.
Tilanteissa, joissa klonaalisten B-solujen immunofenotyyppi poikkeaa tyypillisestä KLL:n immunofenotyypistä, tutkitaan erotusdiagnostisessa mielessä biopsianäytteen histologia ja immunohistokemialliset tutkimukset . CD23 –negatiivisten tai vahvasti kevytketjuilmentymän omaavien tapausten yhteydessä on tutkittava joko cyklin-D1 tai FISH t(11;14) manttelisolulymfooman osoittamiseksi/poissulkemiseksi. CD23 voi olla positiivinen manttelisolulymfoomassa, mutta CD200 on usein negatiivinen.
Luuydinnäytteet
- Diagnoosivaiheessa luuydinnäytteet eivät yleensä ole tarpeen.
Kuvantamistutkimukset
- harkinnan mukaan/ tarvittaessa thoraxröntgen ja vatsan ultraäänitutkimus
Kantasolujensiirto
- Kirjaudu tai rekisteröidy kommentoidaksesi
Kantasolujensiirto
Päivitetty versio tulossa...
Luokittelu
Luokittelu
Kliininen ennusteluokitus: Binet´n luokitus
Binet’n luokitus perustuu affisioituneiden alueiden lukumäärään, hemoglobiinipitoisuuteen sekä trombosyyttiarvoon. Huomioidaan palpoiden onko lymfadenopatia (> 1 cm läpimittainen imusolmuke) uni- vai bilateraalinen.
Levinneisyysalueet:
1) Pää ja kaula, Waldeyerin rengas mukaan lukien, sisältää yhden alueen vaikka useampi imusolmukeryhmä olisi affisioitunut.
2) Kainalot (molemmat kainalot muodostavat yhden alueen)
3) Nivustaipeet, sisältäen pinnalliset reiden alueet (molemmat nivustaipeet muodostavat yhden alueen)
4) Palpoituva perna
5) Palpoituva maksa (kliinisesti suurentunut)
Luokka A (median survival 12 vuotta)
Hb ≥ 100 g/l, trombosyytit ≥ 100 x 109/l ja tautia korkeintaan kahdella alueella
Luokka B (median survival 7 vuotta)
Hb ≥ 100 g/l, trombosyytit ≥ 100 x 109/l ja tautia kolmella tai useammalla alueella.
Luokka C (median survival 2-4 vuotta)
Kaikki potilaat, levinneisyysalueista riippumatta, joilla Hb on < 100 g/l ja/tai trombosyytit < 100 x 109/l
Anemian ja trombosytopenian KLL:aan liittymättömät syyt on selvitettävä ja hoidettava ennen luokitusta. Myös KLL:aan liittyvät immunosytopeniat on hoidettava ennen kuin luokittelu voidaan tehdä.
Luokittelu
- Kirjaudu tai rekisteröidy kommentoidaksesi
Luokittelu
Kliininen ennusteluokitus
Luokitukset perustuvat palpoituvaan lymfadenopatiaan (>1cm), splenomegaliaan ja hepatomegaliaan sekä alentuneisiin verisoluarvoihin. Vain luuydininfiltraation aiheuttamat sytopeniat huomioidaan luokituksessa. Immunosytopeniat ja muut syyt on hoidettava pois ennen luokituksen tekemistä.
Huomioitava, että molemmissa luokituksissa keskimääräiset eliniät tulevat vanhoista tutkimuksista, joissa ei ole käytetty uusia hoitomuotoja.
Binet´n luokitus
Binet’n luokitus perustuu affisioituneiden alueiden lukumäärään, hemoglobiinipitoisuuteen sekä trombosyyttiarvoon. Huomioidaan palpoiden, onko lymfadenopatia (> 1 cm läpimittainen imusolmuke) uni- vai bilateraalinen.
| Binet luokka | Kliiniset löydökset | med. elossaolo |
| A | Hb ≥100 g/l trombosyytit ≥ 100 x10E9/l suurentuneita imusolmukkeita enintään 2:lla alueella |
12v |
| B | Hb ≥ 100 g/l tromb ≥ 100 x 10E9/l 3 tai enemmän affisioituneita imusolmukealueita |
7v |
| C | Hb < 100 ja/ tai tromb < 100 |
2-4 v |
Levinneisyysalueet:
1) Pää ja kaula, Waldeyerin rengas mukaan lukien, sisältää yhden alueen vaikka useampi imusolmukeryhmä olisi affisioitunut.
2) Kainalot (molemmat kainalot muodostavat yhden alueen)
3) Nivustaipeet, sisältäen pinnalliset reiden alueet (molemmat nivustaipeet muodostavat yhden alueen)
4) Palpoituva perna
5) Palpoituva maksa (kliinisesti suurentunut)
Ennustetekijät
Ennustetta huonontavia kliinisiä tai perusverikokein määritettäviä tekijöitä
- Binet- ja RAI-luokka
- Lymfosyyttien kahdentumisaika < 6kk
- Kohonnut seerumin beta-2-mikroglobuliini
- Kohonnut seerumin tymidiinikinaasi
Ennusteeseen vaikuttavia geneettisiä tekijöitä
IGHV-geenin somaattiset hypermutaatiot eli mutaatiostatus:
Ennustetta heikentävänä tekijänä ei-mutatoitunut IGHV-geeni (”unmutated, U- CLL”). Tauti luokittuu ei-mutatoituneeksi silloin kun IgH-uudelleenjärjestymän IGVH-geenin samankaltaisuus on ≥ 98% ns. ituratasekvenssiin verrattuna.
Mutatoituneessa tautimuodossa (”mutated, M-CLL”) IGHV-geenin samankaltaisuus on < 98% ns. ituratasekvenssiin verrattuna
Huom. Mutatoitumisasteeltaan raja-arvoiselle, 97 — < 98%:n alueelle sijoittuvia tapauksia ei kuitenkaan voida yksiselitteisesti luokitella mutatoituneiksi tai ei-mutatoituneiksi.
Stereotypia:
Klonaalisen IGH-uudelleenjärjestymän kuuluminen johonkin kliinisesti merkittävään stereotyyppiluokkaan #1, #2, #4 tai #8. Näistä luokkaan #4 liittyy suotuisa ennuste, muihin taas aggressiivinen taudinkuva (Stamatopoulos ym. Leukemia 2017)
IGHV3-21-geeni klonaalisen IgH-uudelleenjärjestymän IGVH-geeninä on ennustetta huonontava tekijä jos uudelleenjärjestymä samalla luokittuu stereotypialuokkaan #2. Muussa tapauksessa ennuste määräytyy IGHV-geenin mutaatiostatuksen perusteella. (Baliakas ym. Blood 2015).
Sytogenetiikan löydökset:
Kromosomialueen 13q deleetio on yleisin KLL:ssa tavattu poikkeavuus: se on todettavissa n. 30-50 %:lla KLL-potilaista. Ainoana poikkeavuutena tavattuna 13q deleetio liittyy suotuisaan ennusteeseen.
Kromosomin 12 trisomia todetaan n. 15 %:lla KLL-potilaista. Ennuste jonkin verran heikompi kuin normaalin karyotyypin KLL:ssa
Kromosomin 11 q-varren deleetio on todettavissa n. 10-17 %:lla ja kromosomin 17p-varren deleetio 10-15 %:lla KLL-potilaista.
Sekä 11q- että 17p- deleetiot ovat ennustetta huonontavia löydöksiä. Molemmat muutokset voivat syntyä tautisoluihin myös myöhemmin taudin edetessä.
11q- ja 17p- löydöksiä tavataan useammin mutaatiostatukseltaan ei-mutatoituneessa tautimuodossa. Suotuisamman ennusteen muutokset (13q-deleetio ainoana poikkeavuutena) taas ovat yleisempiä mutatoituneessa taudissa.
Kompleksi karyotyyppi liittyy myös KLL:ssa huonon ennusteeseen.
TP53-geeni sijaitsee kromosomialueella 17p ja sen deleetio ja myös muut mutaatiot heikentävät KLL:n ennustetta. 17p-deleetio / TP53-mutaatio ilmaantuu usein uutena muutoksena taudin edetessä. Tällöin poikkeavuuden yleisyys on voi olla 40-50 %:n luokkaa. Valtaosalla 17p-deleetio tapauksista myös toinen alleeli TP53-geenistä on mutatoitunut. TP53-geeni voi olla mutatoitunut myös tapauksissa, joissa kromosomin 17p- deleetiota ei ole ollut osoitettavissa (yleisyys n. 5 %). Näin ollen normaali tulos FISH-tutkimuksessa ei poissulje TP53-mutaation mahdollisuutta.
17p-deleetio ja TP53-mutaatio ovat merkittäviä myös pienen prosenttiosuuden- ja subklonaalisina löydöksinä.
NOTCH1-geenin mutaatioita todetaan n.5- 10 %:lla KLL-potilaista. NOTCH1-mutaatiot assosioituvat +12-trisomiaan ja IGHV-mutaatiostatukseltaan ei-mutatoituneeseen KLL:aan. NOTCH1-mutaatioihin liittyy suurentunut riski taudin transformaatiolle.
SF3B1-geenin mutaatioita esiintyy 10-15 %:lla KLL-potilaista. SF3B1-mutaatiot liittyvät 11q- kromosomideleetioon ja huonoennusteiseen taudinkuvaan.
Seuranta ja vastearvio
Seuranta ja vastearvio
Seuranta
Kontrollikäynnin tai –kontaktin yhteydessä selvitetään aina
- yleisoireet
- infektioiden esiintyminen
- kliininen tutkimus (imusolmukkeet, perna, maksa)
- täydellinen verenkuva
Pitkäaikaista hoitoa (esim BCR- ja BCL-inhibiittorit) saavien potilaiden haittavaikutukset kysellään tarkoin ja niiden ilmaantumisesta pyydetään ilmoittamaan myös sovittujen kontrollien välissä. Siedettävyyden ja tehon perusteella arvioidaan haittavaikutusten oireenmukaisen hoidon, lääkityksen keskeyttämisen tai annosmuutosten tarve. Jatkuvaa hoitoa saavien potilaiden seuranta tapahtuu hematologiaan perehtyneen lääkärin valvonnassa.
Oireetonta ja pienimassaista (Binet A) KLL:aa seurataan avoterveydenhuollossa. Alkuun kontrolli esimerkiksi 3-4 kk kuluttua ja jos tilanne on rauhallinen, voi kontrollit harventaa 6 -12 kk välein tapahtuvaksi.
Seurantaohje potilaalle ja seurannasta vastaavaan yksikköön
Etenevä tai oireinen (Binet B) vaatii tarkempaa seurantaa 2-4 kk välein ja seuranta tapahtuu joko hematologia konsultoiden avopuolella tai hematologiaan perehtyneen lääkärin toimesta.
Määräaikaisten hoitojen jälkeen vastearvio tehdään hoitavassa yksikössä 2 - 6 kk hoitojen päättymisestä. Hoitojen tavoitteesta ja saavutetusta hoitovasteesta riippuen seuranta voi tapahtua myös avopuolella esimerkiksi aluksi 6 -12 kk välein. Saavutettu hoitovaste korreloi yleensä taudin etenemisvapaan ajan kestoon. Oireiden lisääntyessä seurantaa tihennetään esimerkiksi 3-6 kk välein tapahtuvaksi. Hoitoindikaatioiden täyttyessä uudelleen potilas lähetetään hoitoarvioon hematologiseen yksikköön.
Vastearvio
Vaste annetulle hoidolle arvioidaan aikaisintaan 2 kk (vasta-ainehoidot) ja viimeistään 6 kk hoitojen päättymisestä. Jatkuvien hoitojen yhteydessä hoitovaste arvioidaan hoidon aikana, kun on kulunut vähintään 2 kk parhaimman vasteen saavuttamisesta.
Kliinisessä työssä arvioidaan
aina:
- anamneesi (yleisoireet)
- kliininen tutkimus (imusolmukkeet, perna, maksa)
- täydellinen verenkuva
harkinnan mukaan:
- luuydinaspiraatti / -biopsia: pitkittyneet sytopeniat, joskus CR:n varmistamiseksi
- Thx-rtg ja vatsan / imusolmukealueiden UÄ; jos aluksi poikkeava, epäluotettava palpaatio. Vartalon TT vain erityistapauksissa.
Jäännöstautianalyysi veren virtaussytometrialla toistaiseksi vain tutkimusasetelmissa.
Mukailtu taulukko: IWCLL 2008, M. Hallek et al, Blood 15 June 2008, Vol 111, n:o 12, p. 5451 ja iwCLL 2017 Revised Guidelines
|
|
Täydellinen remissio |
Osittainen hoitovaste |
Progressiivinen tauti |
|
A) Tautimassa: |
|
|
|
|
Imusolmukkeet |
ei yli 1,5 cm |
vähintään 50% pieneneminen/vähenemä |
vähintään 50% kasvu |
|
Pernan koko1 |
normaali (kork.13 cm) |
vähintään 50% pienenemä |
vähintään 50% kasvu |
|
Maksan koko2 |
normaali |
vähintään 50% pienenemä |
vähintään 50% kasvu |
|
B-Lymfosyytit3 |
alle 4,0 x 10E9/l |
vähintään 50% lasku lähtötasosta |
vähintään 50% nousu lähtötasosta |
|
Yleisoireet |
ei yleisoireita |
ei määritelty |
ei määritelty |
|
Luuydin4 |
normaali solukkuus, lymf alle 30%, ei B-lymfosyyttinoduluksia |
50% infiltraatioasteen pienenemä tai B-lymfosyyttinoduluksia |
ei määritelty |
|
B) Hematopoieesi: |
|
|
|
|
Hb |
yli 110 g/l (ilman kasvutekijää tai transfuusioita) |
yli 110 tai vähintään 50% nousu lähtötasosta |
vähintään 20 g/l lasku lähtötasosta (KLL:sta johtuva) |
|
Trom |
yli 100 x 10E9/l |
yli 100 tai vähintään 50% nousu lähtötasosta |
vähintään 50% lasku lähtötasosta (KLL:sta johtuva) |
|
Neutr |
yli 1,5 x 10E9/l |
yli 1,5, tai vähintään 50% nousu lähtötasosta |
ei määritelty |
Stable disease, SD = osittaisen hoitovasteen (PR) eikä tautiprogression (PD) kriteeristö ei täyty
1 Splenomegalia yksistään merkityksetön, jos muut CR:n kriteerit täyttyvät (ainakin MRD neg), 2 Paikalliset ja yleistyneet maksanodulukset huomioitava maksan koon lisäksi, 3 BCR-estäjillä hoidettaessa lääkkeeseen liittyvä lymfosytoosi yksistään ei ole puuttuvan hoitovasteen merkki, 4 CR i= CR:n B-kriteeristö jää vajaaksi tai hyposellulaarinen ydin.
Taudin status (SHR-muuttuja)
Taudin status (SHR-muuttuja)
Vastaa SHR-muuttujaa: status-cll
|
Koodi |
Lyhenne |
Nimike |
Määritelmä |
|
0 |
Dg |
Diagnoosivaihe |
Krooninen monoklonaalinen lymfosytoosi (perifeerisen veren B-lymfosyytit > 5,0 x 109/L) ja korkeintaan 55% lymfosyyteistä voi olla atyyppisiä, prolymfosyyttejä tai lymfoblasteja JA B-lymfosyyteillä on tyypillinen immunofenotyyppi: CD19+, CD20+ (heikko), CD23+, CD5+ (ei muita T-solumarkkereita kuten CD2 tai CD3), CD79b (heikko) |
|
1 |
CR-MRDneg |
Remissio, jäännostauti negatiivinen |
Kaikki seuraavista todettavissa aikaisintaan 2 kk hoidon päättymisestä:
|
|
2 |
CR-MRDpos |
Remissio, jäännostauti positiivinen |
Kaikki seuraavista todettavissa aikaisintaan 2 kk hoidon päättymisestä:
|
|
4 |
CR |
Täydellinen vaste (complete remission) |
Kaikki seuraavista todettavissa aikaisintaan 2 kk hoidon päättymisestä:
|
|
5 |
CRi |
Täydellinen vaste, osittainen toipuminen (complete remission, incomplete recovery) |
Kriteerit kuten CR, paitsi:
|
|
6 |
PR |
Osittainen remissio |
Tilan kesto vähintään 2 kuukautta ja todetaan 1.) ≥ 50% pienentyminen veren lymfosyyttilukemassa 2.) ≥ 50% pienentyminen imusolmukkeissa (joko korkeintaan 6 imusolmukkeen läpimittojen summassa tai suurimman imusolmukepaketin läpimitassa), eikä kasvua missään imusolmukkeessa eikä uusia imusolmukesuurentumia. Alle 2 cm läpimittaisten imusolmukkeiden kohdalla alle 25%:n kasvua ei katsota merkittäväksi. 3.) ≥ 50% pienentyminen hepato- tai splenomegaliassa, siltä osin kuin nämä (kohdat 1,2 ja 3) olivat poikkeavia hoidon alkaessa JA 4.) vähintään yksi seuraavista:
|
|
7 |
SD |
Vakaa tauti (stable disease) |
Potilaat, jotka eivät saavuttaneet CR:a tai PR:a ja joilla ei todeta PD:a |
|
8 |
RD |
Hoidelle vastaamaton tauti (refractory disease) |
Kaikki muut paitsi CR, PR tai SD (myös exitus) 6 kk:n kuluessa antileukeemisen hoidon päättymisestä |
|
9 |
PD |
Etenevä tauti (progressive disease) |
Vähintään YKSI seuraavista:
B-Hb:n lasku > 20 g/L tai B-Hb < 100 g/L TAI B-Trombosyyttien lasku > 50% tai B-Trombosyytit < 100 x 109/L, jos antileukeemisen hoidon päättymisestä on kulunut vähintään 3, mutta alle 6 kk, ja luuydinbiopsiassa todetaan klonaalisia KLL-soluja Sytopenioita ei voida käyttää tautiprogression kriteereinä hoidon aikana. |
|
10 |
Relapsi |
Relapsi |
Relapsi voidaan todeta jos potilas on vähintään 6 kk aiemmin saavuttanut CR:n tai PR:n ja todetaan PD |
|
11 |
Relapsi (neuro) |
Isoloitu neurorelapsi |
|
|
90 |
Exitus-relapsi |
Exitus, relapsi |
|
|
91 |
Exitus-infektio |
Exitus, infektio |
|
|
92 |
Exitus-aplasia |
Exitus, aplasia |
|
|
99 |
Exitus-tuntematon |
Exitus, syy tuntematon |
|
|
999 |
Poistunut seurannasta |
Poistunut seurannasta (lost to follow-up) |
|
Kantasolujensiirto
Kantasolujensiirto
Allogeeninen kantasolujensiirto
Indikaatio:
- hyväkuntoinen (60-(65)-v potilas, jolla todettu KLL:n progressio BCR- tai BCL2- inhibiittorin jälkeen ja jolla on sisarusluovuttaja tai hyvä rekisteriluovuttaja
Kirjallisuutta
Dreger P, Ghia P, Schetelig J, van Gelder M, Kimby E, Michallet M, Moreno C, Robak T, Stilgenbauer S, Montserrat E; ERIC and EBMT. High-risk chronic lymphocytic leukemia in the era of pathway inhibitors: integrating molecular and cellular therapies. Blood 2018;132:892-902.
Kirjallisuutta
Kirjallisuutta
- Binet J-L ym. Perspectives on the use of new diagnostic tools in the treatment of chronic lymphocytic leukemia. Blood 2006, 107: 859-861
- Cazin BF ym. Oral Fludarabine Phosphate and Cyclophosphamide in Previously Untreated CLL: Final Response and Follow-Up in 75 Patients. Blood 2003, 102, abst. 1598
- Hallek M ym. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the InternationalWorkshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute–Working Group 1996 guidelines. Blood 2008, 111: 5446
- Damle RN ym. Ig V Gene Mutation Status and CD38 Expression As Novel Prognostic Indicators in Chronic Lymphocytic Leukemia. Blood 1999, 94: 1840-1847
- Döhner H ym. Genomic Aberrations and Survival in Chronic Lymphocytic Leukemia. N Engl J Med 2000, 343: 1910-1916
- Fischer K ym. Bendamustinein Combination with Rituximab (BR) for Patients with Relapsed Chronic Lymphocytic Leukemia (CLL): A Multicentre Phase II Trial of the German CLL Study Group (GCLLSG).Blood (ASH Annual Meeting Abstracts), Nov 2007; 110: 3106.
- Keating MJ ym. Early Results of a Chemoimmunotherapy Regimen of Fludarabine, Cyclophosphamide, and Rituximab As Initial Therapy for Chronic Lymphocytic Leukemia, J Clin Oncol 2005, 23: 4079-4088
- Keating MJ ym. Management guidelines for use of alemtuzumab in B-cell chronic lympho-cytic leukemia. Clin Lymph 2004, 4: 220-227
- Knauf W ym. BendamustineVersus Chlorambucil in Treatment-Naive Patients with B-Cell Chronic Lymphocytic Leukemia (B-CLL): Results of an International Phase III Study.
- Blood (ASH Annual Meeting Abstracts), Nov 2007; 110: 2043.
- Lamanna N ym. Pentostatin, Cyclophosphamide, and Rituximab Is an Active, Well-Tolerated Regimen for Patients With Previously Treated Chronic Lymphocytic Leukemia. J Clin Oncol 2006, 24:1575-1581
- Lundin J ym. Phase II trial of subcutaneous anti-CD52 monoclonal antibody alemtuzumab (Campath-1H) as first-line treatment for patients with B-cell chronic lymphocytic leukemia (B-CLL). Blood 2002, 100: 768-773
- Moreno C ym. Allogeneic Stem-Cell Transplantation May Overcome the Adverse Prognosis of Unmutated VH Gene in Patients With Chronic Lymphocytic Leukemia. J Clin Oncol 2005, 23: 3433-3438
- Moreton P ym. Eradication of Minimal Residual Disease in B-Cell Chronic Lymphocytic Leukemia After Alem-tuzumab Therapy Is Associated With Prolonged Survival. J Clin Oncol 2005, 23: 2971-2979
- Oscier DG ym. Multivariate analysis of prognostic factors in CLL: clinical stage, IGVH gene mutational status, and loss or mutation of the p53 gene are independent prognostic factors. Blood 2002, 100: 1177-1184
- Rawston AC ym. Quantitation of minimal disease levels in chronic lymphocytic leukemia using a sensitive flow cytometric assay improves the prediction of outcome and can be used to optimize therapy. Blood 2001, 98: 29-35
- Schroers R ym. Combined analysis of ZAP-70 and CD38 expression as a predictor of disease progression in B-cell chronic lymphocytic leukemia. Leukemia 2005, 19: 750-758
- Shanafelt ym. Prognosis at diagnosis: integrating molecular biologic insights into clinical practice for patients with CLL. Blood 2004, 103: 1202-1210
- Sorror ML ym. Hematopoietic Cell Transplantation After Nonmyeloablative Conditioning for Advanced Chronic Lymphocytic Leukemia. J Clin Oncol 2005, 23: 3819-3829
- Thursky KA ym. Spectrum of infection, risk and recommendations for prophylaxis and screening among patients with lymphoproliferative disorders treated with alemtuzumab. Br J Haematol 2006, 132: 2-12
- Wierda WG ym. Novel Immune-Based Treatment Strategies for Chronic Lymphocytic Leukemia. J Clin Oncol 2005, 23: 6325-5332
- PettittARym. Alemtuzumab in combination with high-dose methylprednisolone is a logical, feasible and highly active therapeutic regimen in chronic lymphocytic leukemia patients with p53 defects. Leukemia 2006, 20: 1441-1445
Erityiskysymyksiä
- Kirjaudu tai rekisteröidy kommentoidaksesi
Erityiskysymyksiä
KLL ja immuunisytopeniat
Esiintyvyys
- AIHA 4-10%·
- ITP 2-5 %
-
RCA ( punasoluaplasia) n. 1%
-
immuuni neutropenia, harvinainen alle 1%
Sytopenian yhteydessä poissuljettava muut etiologiset tekijät
-
luuydinnäytteen otto usein indisoitu
-
ei randomoituja tutkimuksia hoidosta
Perusperiaate
- Jos KLL muuten rauhallinen, hoida immuunisytopeniaa samoin periaattein kuin potilailla, joilla ei ole KLL
- Jos hoitovastetta ei saada tai se on lyhyt, KLL:n suunnatut hoidot
AIHA
- 4-10%
- prevalenssi korkeampi edenneessä sairaudessa, mutta voi olla myös KLL ensimmäinen esiintymä
- poissulje muut anemian syyt
- diagnoosi
- Hb tason lasku
- hemolyysitutkimukset : Coombs, bilirubiini, e-retik, LD, haptoglobiini
Hoito
A. Ensilinjan hoidot
- Kortisoni
- Predniso(lo) n 1 (- 1,5 ) mg/ kg po
- kunnes hematologinen vaste, asteittainen vähennys
- vasteet tavallisesti 4 viikon kuluessa. Ellei, mieti muuta hoitoa
- Immunoglobuliini-infuusiot
- 0.4 mg/kg / vrk 5 vrk ajan ( vaihtoehto 1g/kg/vrk 1.-2 pv)
- nopeampi, mutta lyhytkestoinen vaste
- esim.lisänä alkutilanteessa kortisonin rinnalla
- punasolujensiirrot
- kliinisen arvion perusteella
- usein alkuvaiheessa tarpeen kohtuullisen Hb tason saavuttamiseksi
- Sopivuuskokeet hankalia, mutta eivät estä siirtoja
- Muista aktiivi yhteistyö veripalvelun kanssa
B.Toisen linjan hoidot
- rituksimabi
- 375mg/m2 iv kerran viikossa x 4
- splenektomia
muista rokotukset edeltävästi
C. Muut hoitovaihtoehdot
- syklosporiini
- 5-8mg/kg jaettuna kahteen annokseen
- tavoite CyA n. 100
- atsatiopriini
- aloitusannos 1-2 mg/kg
- tarvittaessa nosto ad 2.5mg/kg, ellei 4 vkossa vastetta
D. Tukihoidot
- foolihapposubstituutio
Ellei vastetta tai vaste on lyhytaikainen ; arvio KLL- hoitojen aloittaminen!
ITP
- esiintyvyys 2-5%
- erotusdignostiikassa poissujettava muut trombosytopenian aiheuttajat
hoito
- Ensilinjan ja toisen linjan hodot kuten yllä AIHAn kohdalla
- Kortisonihoitona Prednisolonin sijaan voi vaihtoehtoisesti käyttää Dexametason 40mg x 1 po 4 vrk ajan , toistaen joka toinen viikko
- Muut hoitovaihtoehdot
- TPOanalogit
- eltrombopag
- romiplostiimi
- aineistot pieniä, ei tietoa pitkäaikaisvaikutuksista
- TPOanalogit
RCA ( punasoluaplasia )
- diagnoosi perustuu aina luuydinbiopsiaan
- poissulje
- virusinfektiot mm. parvovirus, CMV ja EBV
- EPO vasta-aineet jos potilas on edeltävästi saanut ESA
Hoito
- punasolujensiirrot
- kortisoni
- immuunosuppressiivinen hoito
- käytetyin syklosporiini
- rituksimabi
- alemtutsumabi ( FIMEAN erityislupavalmiste)
Immuunineutropenia
- isoloitu neutropenia ilman muita muutoksia soluarvoissa
- harvinainen, alle 1%
- Hoito
- kortisoni
- G- CSF
Immuunisytopeniat
- Kirjaudu tai rekisteröidy kommentoidaksesi
Immuunisytopeniat
Yleistä
Esiintyvyys
- AIHA 4-10%·
- ITP 2-5 %
-
RCA ( punasoluaplasia) n. 1%
-
immuuni neutropenia, harvinainen alle 1%
Sytopenian yhteydessä poissuljettava muut etiologiset tekijät
-
luuydinnäytteen otto usein indisoitu
-
ei randomoituja tutkimuksia hoidosta
Perusperiaate
- Jos KLL muuten rauhallinen, hoida immuunisytopeniaa samoin periaattein kuin potilailla, joilla ei ole KLL
- Jos hoitovastetta ei saada tai se on lyhyt, KLL suunnatut hoidot
AIHA
- 4-10%
- prevalenssi suurempi edenneessä sairaudessa, mutta voi olla myös KLL ensimmäinen esiintymä
- poissulje muut anemian syyt
- diagnoosi
- Hb tason lasku
- hemolyysitutkimukset : Coombs, bilirubiini, e-retik, LD, haptoglobiini
Hoito
Ensilinjan hoidot
-
kortisoni
-
Predniso(lo) n 1 (- 1,5 ) mg/ kg po
-
kunnes hematologinen vaste, asteittainen vähennys
-
vasteet tavallisesti 4 viikon kuluessa, ellei mieti muuta hoitoa
Immunoglobuliini-infuusiot
- 0.4 mg/kg / vrk 5 vrk ajan ( vaihtoehto 1g/kg/vrk 1.-2 pv)
- nopeampi, mutta lyhytkestoinen vaste
- esim.lisänä alkutilanteessa kortisonin rinnalla
- punasoluinfuusiot
- kliinisen arvion perusteella
- usein alkuvaiheessa tarpeen kohtuullisen Hb tason saavuttamiseksi
- Sopivuuskokeet hankalia, mutta eivät estä siirtoja
- Muista aktiivi yhteistyö veripalvelun kanssa
Toisen linjan hoidot
· rituksimabi
· 375mg/m2 iv kerran viikossa x 4
· splenektomia
· muista rokotukset edeltävästi
Muut hoitovaihtoehdot
· syklosporiini
· 5-8mg/kg jaettuna kahteen annokseen
· tavoite CyA n. 100
· atsatiopriini
· aloitusannos 1-2 mg/kg
· tarvittaessa nosto ad 2.5mg/kg ,jos 4 viikossa ei vastetta
· Tukihoidot
· Foolihapposubstituutio
- Ellei vastetta tai vaste on lyhytaikainen ; arvio KLL- hoitojen aloittaminen!
ITP
- esiintyvyys 2-5%
- erotusdignostiikassa poissujettava muut trombosytopenian aiheuttajat
- hoito
* Ensilinjan ja toisen linjan hodot kuten yllä AIHAn kohdalla
Kortisonihoitona Prednisolonin sijaan voi vaihtoehtoisesti käyttää Dexametason 40mg po 4 vrk, toistaen joka toinen viikko
* Muut hoitovaihtoehdot
· TPOanalogit
· eltrombopag
· romiplostiimi
· aineistot pieniä, ei tietoa pitkäaikaisvaikutuksista
RCA ( punasoluaplasia )
- diagnoosi perustuu aina luuydinbiopsiaan
- poissulje virusinfektiot mm. parvovirus, CMV ja EBV
- poissulje myös EPO vasta-aineet jos potilas on edeltävästi saanut ESA
- Hoito
· punasolusiirrot
· kortisoni
· immuunosuppressiivinen hoito
· käytetyin syklosporiini
· rituksimabi
· alemtutsumabi ( FIMEAN erityislupavalmiste)
Immuunineutropenia
- isoloitu neutropenia ilman muita muutoksia soluarvoissa
- harvinainen, alle 1%
- Hoito
· kortisoni
· G- CSF
Infektiot
- Kirjaudu tai rekisteröidy kommentoidaksesi
Infektiot
Infektioiden ehkäisy kroonisessa lymfaattisessa leukemiassa
KLL:n hoito valitettavan harvoin korjaa tautiin liittyvää immuunipuutosta. Sen sijaan KLL:n hoito voi – ainakin väliaikaisesti - heikentää immuunipuolustusta. Lääkkeillä voidaan estää herpesinfektioita (asikloviiri, valasikloviiri), pneumocystis jirovecii (PCP)- ja toksoplasmainfektioita (sulfametoksatsoli-trimetopriimi) sekä hepatiitin aktivoitumista (lamivudiini). Bakteeriantibioottiprofylaksiaa ei rutiinisti suositella.
Antimikrobiprofylaksiaa suositellaan seuraavissa tilanteissa:
- kortikosteroidiannos, joka vastaa > 20 mg/vrk Prednisolonia yli kuukauden ajan
- alemtutsumabihoito
- FCR-hoito, bendamustiinihoito. Erityisesti, jos potilaalle ilmaantuu hoidon seurauksena lymfopeniaa (< 1.0 x 109/l), pitkittynyttä neutropeniaa, ikä on > 65v tai on pitkälle edennyt tauti. Aina ei ensilinjan hoidossa ole tarpeen.
- potilas on aiemmin sairastanut pneumocystis-keuhkokuumeen tai anamneesissa on toistuvia herpestulehduksia.
- veren CD4 solut ovat < 0.2 x 109/l
- jos hoitoon tiedetään liittyvän > 3.5% riski PCP-infektioon.
- jos potilas on toksoplasma –seropositiivinen ja CD4+ solut < 0.1 x 109/l, on sulfa-trimetopriimi syytä antaa toksoplasma-annoksella (kts alla)
- jos potilas on hepatiitti-B:n kantaja (HBsAg +, HBV-DNA+)
Mitä ehkäistään ja millä:
- Pneumocystis jirovecii (PCP):
- Sulfametoksatsoli-trimetropriimi (Cotrim *) 1 x 1 joka pv (vaihtoehtoisesti Cotrim forte 1 x1 kolmasti viikossa.)
Huom! Ditrim on sulfadiatsiinia, sen tehosta ei ole näyttöä - Pentamidiini inhalaatiot 100mg 4 viikon välein
- Dapsoni 100 mg x 1 tai atovakoni 1500 mg x 1 joka pv
- Sulfametoksatsoli-trimetropriimi (Cotrim *) 1 x 1 joka pv (vaihtoehtoisesti Cotrim forte 1 x1 kolmasti viikossa.)
- Herpes simplex, varizella zoster:
- Asikloviiri 400mg (800mg) x 2, valasikloviiri 250-500 mg x 2
- Toksoplasma gondii:
- Cotrim forte 1 x 1 joka päivä
- Sytomegalovirus (CMV):
- Alemtutsumabihoidossa suositellaan seuraamaan CMV nukleiinihapon määrää viikoittain. Jos potilaalle tulee oireinen CMV-infektio, alemtutsumabi keskeytetään ja hoidetaan 2-3 viikkoa (val)gansikloviirillä. Alemtutsumabihoito voidaan aloittaa uudelleen kun potilas on oireeton ja CMV-PCR negatiivinen. Oireetonta CMV-reaktivaatiota joko ei hoideta tai vaihtoehtoisesti 1-2 viikkoa hoidetaan, mutta alemtutsumabia saa silloin jatkaa. (Hoitopäätös potilaskohtaisesti, herkästi infektiolääkärin konsultaatio).
- Hepatiitti B:
- Infektiolääkärin konsultaatio.Lamivudiini 100mg x1
Kuinka kauan profylaksiaa jatketaan:
2-6 kk hoidon päättymisestä tai niin kauan kun CD4-solut < 0.2 x 109/l
Immunoglobuliini
Pelkän matalan seerumin immunoglobuliinitason takia ei ole syytä aloittaa immunoglobuliinihoitoa. Hoito on perusteltua, jos seerumin IgG < 3 g/l ja potilaalla on ollut > 2 bakteeri-infektiota edeltävän vuoden aikana.
Iv-annos n. 400 mg / kg 3-4 viikon välein ja tavoitteena on infektioiden häviäminen ja normaali IgG-pitoisuus ennen seuraavaa infuusiota. (Käytetty myös pienempiä annoksia, ruotsalaisten suosituksessa vakioannos 10 g 3 viikon välein.) Mahdollista antaa myös subkutaanisesti (annos n. 50 mg/kg/viikko). Jos infektiot jatkuvat riittävästä substituutiosta huolimatta, hoito lopetetaan.
Rokotukset
Pneumokokkikonjugaatti kaikille mahdollisimman varhaisessa vaiheessa (+/- polysakkaridirokote 2 kk myöhemmin, jos erityisen suuri riski). Influenssarokote syksyisin, syytä antaa myös perheenjäsenille.
Eläviä rokotteita syytä välttää(BCG, oraalinen polio, MPR, salmonella, keltakuume, vesirokko, rotavirus, oraalinen lavantauti). Rokotteita myös syytä välttää aktiivisessa autoimmuunihemolyysivaiheessa, saattaa pahentaa tilannetta.
Hoito
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hoito
Hoidon tavoitteeseen vaikuttaa mm. potilaan kunto ja oletettavissa oleva KLL:sta riippumaton eliniän ennuste. Hyväkuntoisilla (Fit) potilailla hoidon tavoite on täydellinen remissio, jonka on todettu parantavan tautivapaata aikaa ja kokonaiselinaikaa. Huonompikuntoisilla ja iäkkäillä (Slow go) tyydytään oirekontrolliin ja pyritään estämään taudin eteneminen. Hyvin haurailla potilailla tyydytään hyvään oireenmukaiseen hoitoon.
Ennen hoidon aloitusta on tärkeää arvioida potilaan kokonaistilannetta ja kykyä sietää immuno- ja myelosuppressiivista hoitoa. Potilaaseen liittyviä hoidonvalintaan vaikuttavia tekijöitä ovat mm. fyysinen suorituskyky, toimintakyky, ikä, liitännäissairaudet ja niiden hoitoon käytettävät lääkkeet. Myös valittavaan lääkitykseen liittyvät haitta- ja yhteisvaikutukset, potilaan oma mielipide ja valmius sitoutua valittavaan hoitoon on huomioitava hoitoa suunniteltaessa. Jatkuvaksi tarkoitettu lääkehoito keskeytetään / vaihdetaan toiseen taudin edetessä hoidon aikana tai jos hoitoon liittyy hallitsemattomia tai vakavia haittavaikutuksia.
Tärkeimpänä tautiin liittyvänä hoidon valintaan vaikuttavana geneettisenä tekijänä on mahdollinen TP53- geenin toiminnanvajaus (Del(17p) tai TP53 -mutaatio). Myös IGHV-geenin mutaatiostatus vaikuttaa hoidonvalintaan, sillä ei-mutatoituneen tautimuodon on todettu vastaavan huonommin kemoimmunoterapialle, myös ilman todettua TP53-geenin toiminnanvajetta. Vastaavasti mutatoituneessa tautimuodossa ilman TP53-geenin toiminnanvajeeseen johtavia muutoksia voidaan kemoimmunoterapialla saavuttaa pitkäkestoisia remissioita. Kemoimmunoterapiaa ei näissä korkeamman riskin taudeissa suositella, mikäli muita hoitovaihtoehtoja on käytettävissä (korvattavuus).
Arvioi aina, soveltuuko potilas meneillään oleviin hoitotutkimuksiin. Alla listattuna niitä hoitovaihtoehtoja annosteluohjeineen, joilla päivityshetkellä KELA-korvattavuus. Listauksen järjestys ryhmien sisällä ei välttämättä ole ensisijaisuusjärjestys, sillä hoidon valinta tapahtuu lopulta yksilöllisesti kaikki em. asiat huomioiden.
Hyväkuntoisten (fit) hoito
Ensilinjan hoito hyväkuntoisille potilaille, joilla ei ole todettu del 17p/TP53mut
Kemoimmunoterapiavaihtoehdot:
FCR
- rituksimabi C1D0: 375 mg/m2 , C2-6D1: 500mg/m2 iv
- fludarabiini C1-6D1-5: 30 mg/m2 po
- syklofosfamidi C1-6D1-5: 200 mg/m2 po
vaihtoehtoisesti
- rituksimabi C1D0: 375 mg/m2 iv , C2-6D1: 500mg/m2 iv
- fludarabiini C1-6D1-3: 40 mg/m2 po
- syklofosfamidi C1-6D1-3: 250 mg/m2 po
harvoin erityistilanteissa (FC i.v.)
- rituksimabi C1D0: 375 mg/m2 iv , C2-6D1: 500mg/m2 iv
- fludarabiini C1-6D1-3: 25 mg/m2 iv
- syklofosfamidi C1-6D1-3: 250 mg/m2 iv
Syklin pituus 28 päivää. Tavoitteena on kuusi hoitosykliä.
AIHA on vasta-aihe fludarabiinille (Coombs positiivisuuden lisäksi merkkejä hemolyysistä). Seuraa hemolyysikokeita hoitojen aikana. Fludarabiinin takia verituotteet annetaan sädetettyinä.
Yli 65-vuotiaille hyväkuntoisille potilaille suositellaan BR-hoitoa, koska FCR-hoidon toksisuus on huomattava tässä ikäryhmässä. Tämän lisäksi se on suositeltavampi potilaille, joilla on munuaisten vajaatoimintaa (Krea-Cl on alle 60 ml/min).
Bendamustiini + rituksimabi (BR)
- rituksimabi
- C1D0: 375 mg/m2 iv
- C2-6D1: 500 mg/m2 iv
- bendamustiini
- C1-6D1-2: 90 mg/m2 iv
Syklin pituus 28 päivää. Tavoitteena kuusi hoitosykliä.
Kemovapaat vaihtoehdot:
Obinututsumabi + venetoklaksi
(Huom! Tutkimusnäyttö toistaiseksi vain unfit-potilasryhmässä)
- obinututsumabi
- C1: D1 100 mg i.v. ja D2 900 mg i.v.
- C1D8 ja D15: 1000 mg i.v.
- C2-6D1: 1000 mg i.v.
- venetoklaksi
- C1D22 - C2D28: annostitraus (20 mg - 50 mg - 100 mg - 200 mg - 400 mg)
- C3-12: 400 mg x1 p.o.
- 12 hoitojakson määräaikainen hoito
Syklin pituus 28 päivää. Tuumorilyysioireyhtymän ehkäisytoimet valmisteyhteenvedon ohjeen mukaisesti
Ensilinjan hoito hyväkuntoisille potilaille, joilla on todettu del 17p/TP53mut
Ibrutinibi
- Ibrutinibi
- 420 mg x1 p.o.
- jatkuva hoito
Huomioi lisääntynyt riski vuotohäiriöihin ja rytmihäiriöihin sekä mahdollinen verenpaineen nousu
Obinututsumabi + venetoklaksi
- obinututsumabi
- C1: D1 100 mg i.v. ja D2 900 mg i.v.
- C1D8 ja D15: 1000 mg i.v.
- C2-6D1: 1000 mg i.v.
- venetoklaksi
- C1D22 - C2D28: annostitraus (20 mg - 50 mg - 100 mg - 200 mg - 400 mg) 5 viikkoa
- C3-12: 400 mg x1 p.o.
- 12 hoitojakson määräaikainen hoito
Syklin pituus 28 päivää. Tuumorilyysioireyhtymän ehkäisytoimet valmisteyhteenvedon ohjeen mukaisesti
Venetoklaksi
- annostitraus 20 mg - 50 mg - 100 mg - 200 mg - 400mg x1 p.o. valmisteyhteenvedon ohjeen mukaisesti
- jatkuva hoito
- jos B-solureseptoriestäjähoito ei sovellu tai se on epäonnistunut
Huomioi tuumorilyysioireyhtymän ehkäisy, ks.valmisteyhteenveto
Idelalisibi + rituksimabi
- idelalisibi
- 150 mg x2 p.o
- jatkuva hoito
- rituksimabi
- 1.infuusio 375 mg/m2
- 500 mg/m2 iv joka 2.viikko (4 infuusiota)
- 500 mg/m2 iv joka 4.viikko (3 infuusiota)
Ks. idelalisibin valmisteyhteenvedosta infektioprofylaksia ja muut seurantaohjeet sekä toimintaohjeet haittavaikutustilanteissa..
Relapoituneen/refraktaarin KLL:n hoito
Edellinen hoito voidaan uusia, mikäli ensilinjan hoidolla saavutettiin yli 3 vuoden kestoinen remissio. Ellei potilaalla ole jo aiemmin tiedossa olevaa TP53-geenin dysfunktiota, suositellaan tutkittavaksi uudelleen verestä FISH-tutkimus (ja B-TP53-D). Mikäli edellisen hoidon päättymisestä on alle 3 vuotta tai kyseessä on refraktaari KLL, hoitoa vaihdetaan. Katso myös kappale Kantasolujensiirto.
Ibrutinibi
- 420 mg x1 p.o.
- jatkuva hoito
Huomioi lisääntynyt riski vuoto- ja rytmihäiriöihin sekä mahdollinen verenpaineen nousu
Venetoklaksi + rituksimabi
- venetoklaksin
- annostitraus 400 mg:aan x1 ja tuumorilyysioireyhtymän ehkäisy kuten venetoklaksi-monoterapiassa
- 2 vuoden määräaikainen hoito
- rituksimabi
- 4 viikon välein aloitetaan vasta venetoklaksin annosnostovaiheen päätyttyä kun potilas käyttänyt 7 vrk:n ajan annosta 400 mg x1
- 1. infuusio 375 mg/m2 i.v.
- 2.-6. infuusio 500 mg/m2 i.v.
Venetoklaksi
- annostitraus 400 mg:aan x1 p.o. valmisteyhteenvedon ohjeen mukaisesti
- jatkuva hoito
- jos B-solureseptoriestäjähoito ei sovellu tai se on epäonnistunut
Huomioi tuumorilyysioireyhtymän ehkäisy, ks. valmisteyhteenveto
Idelalisibi + rituksimabi
- idelalisibi
- 150 mg x2 p.o.
- jatkuva hoito
- rituksimabi
- 1.infuusio 375 mg/m2
- 500 mg/m2 iv joka 2.viikko (4 infuusiota)
- 500 mg/m2 iv joka 4.viikko (3 infuusiota)
Ks. idelalisibin valmisteyhteenvedosta infektioprofylaksia ja muut seurantaohjeet sekä toimintaohjeet haittavaikutustilanteissa.
Arvioi soveltuuko potilas meneillään oleviin hoitotutkimuksiin.
Kirjallisuutta:
Bouvet E, Borel C, Obéric L, Compaci G, Bruno Cazin B, A-S, Guy Laurent G, Ysebaert L. Impact of dose intensity on outcome of fludarabine, cyclophosphamide, and rituximab regimen given in the first-line therapy for chronic lymphocytic leukemia. Haematologica 2013;98:65-70.
Byrd J, Furman R, Coutre S, Flinn I, Burger J, Kristie A. Blum K, Grant B, Sharman J, Coleman M, Wierda W, Jones J, Zhao W, Heerema N, Johnson A, Sukbuntherng J, Chang B, Clow F, Hedrick E, Buggy J, James D, O'Brien S. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N Eng J Med NEJM 2014;369:32-42.
Eichhorst B, Fink A-M, Busch R, Lange E, Köppler H, Kiehl M, Sökler M, Schlag R, Vehling-Kaiser U, Köchling G, Plöger C, Gregor M, Plesner T, Trneny M, Fischer K, Döhner H, Kneba M, Wendtner C, Klapper W, Kreuzer K-A, Stilgenbauer S, Böttcher S, Hallek M. Chemoimmunotherapy With Fludarabine (F), Cyclophosphamide (C), and Rituximab (R) (FCR) Versus Bendamustine and Rituximab (BR) In Previously Untreated and Physically Fit Patients (pts) With Advanced Chronic Lymphocytic Leukemia (CLL): Results Of a Planned Interim Analysis Of The CLL10 Trial, An International, Randomized Study Of The German CLL Study Group (GCLLSG). Proc ASH 2014; Abstract 19.
Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P, Langerbeins P, von Tresckow J, Engelke A, Maurer C, Kovacs G, Herling M, Tausch E, Kreuzer K-A, Eichhorst B, B ¨ottcher S, Seymour JF, Ghia P, Marlton P, Kneba M, Wendtner C-M, D ¨ohner H, Stilgenbauer S and Mic Hallek M. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood 2016;127:208-215
Furman R, Sharman J, Coutre S, Cheson B, Pagel J, Hillmen P, Barrientos J, Zelenetz A, Kipps T, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt A, Ma S, Stilgenbauer S, Cramer P, Aiello M, Johnson D, Miller L, Li D, Jahn T, Dansey R, Hallek M, O'Brien S. Idelalisib and Rituximab in Relapsed Chronic Lymphocytic Leukemia. N Eng J Med 2014;370:997-1007.
Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848-1854
Hallek M, Cheson B, Catovsky D, Caligaris-Cappio F, Guillaume Dighiero G, Hartmut Döhner H, Peter Hillmen P, Keating M, Montserrat E, Rai K, Kipps T. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute–Working Group 1996 guidelines. Blood 2008;12:5446-5456.
Hallek M, Fischer K, Fingerle-Rowson G, Fink A, Busch R, Mayer J, Hensel M, Hopfinger G, Hess G, von Grünhagen U, Bergmann M, Catalano J, Zinzani P, Caligaris-Cappio F, Seymour J, Berrebi A, Jäger U, Cazin B, Trneny M, Westermann A, Wendtner C, Eichhorst B, Staib P, Bühler A, Winkler S, Zenz T, Böttcher S,Ritgen M, Mendila M, Kneba M, Döhner H, Stilgenbauer S, on behalf of an international group of investigators and the German Chronic Lymphocytic Leukaemia Study Group. Addition of rituximab to fl udarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164-1174.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating M, Montserrat E, Chiorazzi N, Stilgenbauer S. Rai KR, Byrd JC, Eichhorst B, O´Brien S, Robak T, Seymor JF, Kipps TJ. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745-2760.
Kater AP, Seymour JF, Hillmen P, Eichhorst B, Langerak AW, Owen C, Verdugo M, Wu J, Punnoose EA, Jiang Y, Wang J, Boyer M, Humphrey K, Mobasher M, and Kipps TJ. Fixed Duration of Venetoclax-Rituximab in Relapsed/Refractory Chronic Lymphocytic Leukemia Eradicates Minimal Residual Disease and Prolongs Survival: Post-Treatment Follow-Up of the MURANO Phase III Study
J Clin Oncol 2019 37:4, 269-277
Seymour JF, Kipps TJ, Eichhorst B, Hillmen P et al. Venetoclax-Rituximab in relapsed or refractory chronic lymphocytic leukemia. NEJM 2018; 378:1107-1120
Stilgenbauer S, Zenz T, Winkler D, Bühler A, Schlenk R F, Groner S, Busch R, Hensel M, Dührsen U, Finke J, Dreger P, Jäger U, Lengfelder E, Hohloch K, Söling U, Schlag R, Kneba M, Hallek M, Döhner H. Subcutaneous Alemtuzumab in Fludarabine-Refractory Chronic Lymphocytic Leukemia: Clinical Results and Prognostic Marker Analyses From the CLL2H Study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2009;24:3994-4001.
Slow go
1.linjan hoito, joilla ei ole todettu del 17p/TP53mut
Obinututsumabi + klorambusiili
- obinututsumabi
- C1: D1 100 mg, D2 900 mg,
- C1D8 ja D15: 1000 mg iv
- C2-6D1: 1000 mg iv
- varauduttava infuusioreaktioihin, kts. esilääkitys valmisteyhteenvedosta
- klorambusiili
- C1-6D1 ja D15: 0.5 mg/kg p.o.
Sykli 28 vrk
Obinututsumabi + venetoklaksi (Obi-Ven)
- obinututsumabi
- C1: D1 100 mg i.v. ja D2 900 mg i.v.
- C1D8 ja D15: 1000 mg i.v.
- C2-6D1: 1000 mg i.v.
- venetoklaksi
- C1D22 - C2D28: annostitraus (20 mg - 50 mg - 100 mg - 200 mg - 400 mg) 5 viikkoa
- C3-12: 400 mg x1 p.o.
- 12 hoitojakson määräaikainen hoito
Syklin pituus 28 päivää. Tuumorilyysioireyhtymän ehkäisytoimet valmisteyhteenvedon ohjeen mukaisesti
Rituksimabi + bendamustiini
- rituksimabi
- C1D0: 375 mg/m2 iv,
- C2-6D1: 500 mg/m2 iv
- bendamustiini
- C1-6D1-2: 70 - 90 mg/m2 iv
Sykli 28 vrk
Rituksimabi + klorambusiili
(vaihtoehtoinen annostelu suluissa; useita erilaisia annosohjeita)
- rituksimabi
- C3D1: 375 mg/m2 iv pv, C4-8D1:500 mg/m2 iv (tai C1D1 375 mg/m2 ja C2-6D1 500 mg/m2)
- klorambusiili
- C1-6D1-7: 8 mg/m2 (tai C1-6D1-7:10mg/m2)
Sykli 28 vrk
1. linjan hoito, joilla on 17p –deleetio/TP53 -mut
Ibrutinibi
- ibrutinibi
- 420 mg x1 p.o.
- jatkuva hoito
Huomioi lisääntynyt riski vuotohäiriöihin sekä rytmihäiriöihin sekä mahdollinen verenpaineen nousu
Obinututsumabi + venetoklaksi
- obinututsumabi
- C1: D1 100 mg i.v. ja D2 900 mg i.v.
- C1D8 ja D15: 1000 mg i.v.
- C2-6D1: 1000 mg i.v.
- venetoklaksi
- C1D22 - C2D28: annostitraus (20 mg - 50 mg - 100 mg - 200 mg - 400 mg) 5 viikkoa
- C3-12: 400 mg x1 p.o.
- 12 hoitojakson määräaikainen hoito
Syklin pituus 28 päivää. Tuumorilyysioireyhtymän ehkäisytoimet valmisteyhteenvedon ohjeen mukaisesti
Venetoklaksi
- annostitraus 400 mg:aan x1 p.o. valmisteyhteenvedon ohjeen mukaisesti
- jatkuva hoito
- jos B-solureseptoriestäjähoito ei sovellu tai se on epäonnistunut
Huomioi tuumorilyysioireyhtymän ehkäisy, ks. valmisteyhteenveto
Idelalisibi + rituksimabi
- idelalisibi
- 150 mg x2 p.o.
- jatkuva hoito
- rituksimabi
- 1.infuusio 375 mg/m2
- 500 mg/m2 joka 2.viikko (4 infuusiota)
- 500 mg/m2 joka 4.viikko (3 infuusiota)
Kts.idelalisibin valmisteyhteenvedosta tarvittava infektioprofylaksia ja muut seurantaohjeet sekä toimintaohjeet haittavaikutustilanteissa.
Relapsin hoito
- hoitovaste kestänyt yli 3 vuotta: edellinen hoito voidaan uusia
- hoitovaste kestänyt alle 3 vuotta: hoidon vaihto
- FCR-hoitojen toistamista ei kuitenkaan suositella kumulatiivisiin annoksiin liittyvän lisääntyneen MDS-riskin vuoksi
Ibrutinibi
- Ibrutinibi
- 420 mg x1 p.o.
- jatkuva hoito
Huomioi lisääntynyt riski vuotohäiriöihin sekä rytmihäiriöihin sekä mahdollinen verenpaineen nousu
Rituksimabi + Venetoklaksi
-
venetoklaksi
-
annostitraus 400 mg:aan x1 ja tuumorilyysioireyhtymän ehkäisy kuten venetoklaksi-monoterapiassa
-
-
rituksimabi
-
aloitetaan vasta venetoklaksin annosnostovaiheen päätyttyä kun potilas käyttänyt 7 vrk:n ajan annosta 400 mg x1
-
C2D8: 1. infuusio 375 mg/m2 i.v.
-
2.-6. infuusio 500 mg/m2 i.v., 4 viikon välein
-
Venetoklaksi
- annostitraus 400 mg:aan x1 p.o. valmisteyhteenvedon ohjeen mukaisesti
- jatkuva hoito
- jos B-solureseptoriestäjähoito ei sovellu tai se on epäonnistunut
Huomioi tuumorilyysioireyhtymän ehkäisy, ks. valmisteyhteenveto
Idelalisibi + rituksimabi
- idelalisibi
- 150 mg x2 p.o.
- jatkuva hoito
- rituksimabi
- 1.infuusio 375 mg/m2
- 500 mg/m2 joka 2.viikko (4 infuusiota)
- 500 mg/m2 joka 4.viikko (3 infuusiota)
Kts.idelalisibin valmisteyhteenvedosta infektioprofylaksia ja muut seurantaohjeet sekä toimintaohjeet haittavaikutustilanteissa.
Kirjallisuutta:
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, O´Brien S. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32-42.
Cortelezzi A, Sciume M, Liberati AM, Vincenti D, Cuneo A, Reda G, Laurenti L, Zaja F, Marasca R, Chiarenza A, Gritti G, Orsucci L, Storti S, Angelucci E, Cascavilla N, Gobbi M, Mauro FR, Morabito F, Fabris S, Piciocchi A, Vignetti M, Neri A, Rossi D, Giannarelli D, Guarini A, Foa R. Bendamustine in combination with Ofatumumab in relapsed or refractory chronic lymphocytic leukemia: a GIMEMA Multicenter Phase II Trial. Leukemia 2014;28:642-8.
Fischer K, Cramer P, Busch R, Böttcher S, Bahlo J, Schubert J, Pfluger K, Schott S, Goede V, Isfort S, von Tresckow J, Fink A-M, Buhler A, Winkler D, Kreuzer K-A, Staib P, Ritgen M, Kneba M, Dohner H, Eichhorst BF, Hallek M, Stilgenbauer S, Wendtner C-M. Bendamustine in combination with rituximab for previously untreated patients with chronic lymphocytic leukemia: A multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2012;30:3209-16.
Fischer K, Cramer P, Busch R, Stilgenbauer S, Bahlo J, Schweighofer CD, Bottcher S, Staib P, Kiehl M, Eckart MJ, Kranz G, Goede V, Elter T, Buhler A, Winkler D, Kneba M, Dohner H, Eichhorst BF, Hallek M, Wendtner C-M. Bendamustine combined with rituximab in patients with relapsed and/or refractory chronic lymphocytic leukemia: A multicenter phase II trial of German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2011;29:3559-66.
Fischer K, Othman A, Bahlo J, Fink A-M, Tandon M, Dixon M, Robrecht S, Warburton S, Humhrey K, Samoylova O, Liberati A, Pinilla-Ibarz J, et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N Engl J Med 2019; 380:2225-2236
Foa R, Del Giudice I, Cuneo A, Del Poeta G, Ciolli S, Di Raimondo F, Lauria F, Cencini E, Matteo Rigolin G, Cortelezzi A, Nobile F, Callea V, Brugiatelli M, Massaia M, Molica S, Trentin L, Rizzi R, Specchia G, Di Serio F, Orsucci L, Ambrosetti A, Montillo M, Luigi Zinzani P, Ferrara F, Morabito F, Angela Mura M, Soriani S, Peragine N, Tavolaro S, Bonina S, Marinelli M, Stefania De Propris M, Della Starza I, Piciocchi A, Alietti A, Runggaldier EJ, Gamba E, Romana Mauro F, Chiaretti S, Guarini A. Chlorambucil plus rituximab with or without maintenance rituximab as first-line treatment for elderly chronic lymphocytic leukemia patients. Am J Hematol 2014 Jan 11. doi:10.1002/ajh.23668 [Epud ahead of print].
Foon KA, Boyiadzis M, Land SR, Marks S, Raptis A, Pietragallo L, Meisner D, Laman A, Sulecki M, Butchko A, Schaefer P, Lenzer D, Tarhini A. Chemoimmunotherapy with low-dose fludarabine and cyclophosphamide and high dose rituximab in previously untreated patients with chronic lymphocytic leukemia. J Clin Oncol 2009;27:498-503.
Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, Ma S, Stilgenbauer S, Cramer P, Aiello M, Johnson DM, Miller LL, Li D, Jahn TM, Dansey RD, Hallek M, O´Brien SM. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 2014;370:997-1007.
Goede V, Fischer K, Busch R, Engelke A, Eichhorst B, Wendtner CM, Chagorova T, de la Serna J, Dilhuydy MS, Illmer T, Opat S, Owen CJ, Samoylova O, Kreuzer KA, Stilgenbauer S, Döhner H, Langerak AW, Ritgen M, Kneba M, Asikanius E, Humphrey K, Wenger M, Hallek M. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014;370:1101-10.
Hallek M. Chronic lymphocytic leukemia: 2013 Update on diagnosis, risk stratification and treatment. Am J Hematol 2015;90:446-60.
Hillmen P, Robak T, Janssens A, Babu KG, Kloczko J, Grosicki S, Doubek M, Panagiotidis P, Kimby E, Schuh A, Pettitt AR, Boyd T, Montillo M, Gupta IV, Wright O, Dixon I, Carey JL, Chang CN, Lisby S, McKeown A, Offner F; COMPLEMENT 1 Study Investigators. Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): a randomised, multicentre, open-label phase 3 trial. Lancet 2015;385:1873-83.
O´Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA, Grant B, Richards DA, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Izumi R, Hamdy A, Chang BY, Graef T, Clow F, Buggy JJ, James DF, Byrd JC. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol 2014;15:48-58.
Wierda WG, Kipps TJ, Mayer J, Stilgenbauer S, Williams CD, Hellmann A, Robak T, Furman RR, Hillmen P, Trneny M, Dyer MJ, Padmanabhan S, Piotrowska M, Kozak T, Chan G, Davis R, Losic N, Wilms J, Russell CA, Osterborg A; Hx-CD20-406 Study Investigators. Ofatumumab as single-agent CD20 immunotherapy in fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol 2010;28:1749-55.
Kansainvälisiä hoitosuosituksia:
Saksa 2020: Chronische Lymphatische Leukämie (CLL) — Onkopedia
Ruotsi 2020: Nationellt vårdprogram Kronisk lymfatisk leukemi (KLL) (cancercentrum.se)
ESMO 2020: Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up (annalsofoncology.org)
NCCN 2019: https://www.nccn.org/professionals/physician_gls/default.aspx
BSH 2018: https://onlinelibrary.wiley.com/doi/epdf/10.1111/bjh.15460
iwCLL 2018: https://doi.org/10.1182/blood-2017-09-806398
Ennen hoitoa
- Kirjaudu tai rekisteröidy kommentoidaksesi
Ennen hoitoa
Otsikko vaihdettiin muodosta <em class="placeholder">Ennen hoitoa</em> muotoon <em class="placeholder">Ennen hoitoa</em>.
Hoidon indikaatiot
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hoidon indikaatiot
Otsikko vaihdettiin muodosta <em class="placeholder">Hoidon indikaatiot</em> muotoon <em class="placeholder">Hoidon indikaatiot</em>.
Hoidon indikaatiot
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hoidon indikaatiot
- Oireetonta Binet A ja B-luokan tautia voidaan usein seurata.
- Koska valkosolujen aggregaatioon liittyviä oireita harvemmiin tulee KLL -potilaille,
ei absoluuttista lymfosyyttitasoa pidetä yksittäisenä hoidon aloituksen aiheena
Hoidon aloituksen indikaatiot
- IWCLL suositus
1. Etenevä luuytimen toiminnan heikkeneminen:
- paheneva anemia ja/tai trombosytopenia
2. Huomattava splenomegalia
- perna ainakin 6 cm kylkikaaren alapuolelle
- etenevä tai oireinen splenomegalia
3. Huomattava lymfadenopatia
- imusolmukkeen läpimitta > 10 cm
- etenevä tai oireinen lymfadenopatia
4. Etenevä veren absoluuttinen lymfosytoosi
- yli 50% nousu 2 kk aikana tai
- lymfosyyttien määrän kaksinkertaistuminen ( LDT) 6 kk aikana
- LDT : Ei tilanteissa, joissa alkuvaiheen lymf < 30 x10E9/l
5. Huonosti kortikosteroidille tai muulle käypä hoidolle reagoiva AIHA tai ITP
6. Tautiin liittyvät yleisoireet
- tahaton vähintään 10% painonlasku viimeisen 6 kk aikana
- merkittävänä väsymys ( ECOG 2 tai huonompi)
- kuumetta vähintään 38.0 ainakin 2 vkon ajan ilman osoitusta infektiosta
- vähintänä 1 kk ajan esiintynyt yöhikoilu
.
Tutkimukset ennen hoitoa
- Kirjaudu tai rekisteröidy kommentoidaksesi
Tutkimukset ennen hoitoa
Laboratoriokokeet
- B-PVK+TKD
- E-Retik
- B-La
- P-CRP
- P-K, P-Na
- P-Krea
- P-Uraat
- P-Gluk
- P-ALAT, P-AFOS, P-Bil
- P-LD
- P-Ca-albk/P-Ca-ion
- E-ABORh
- E-Coomb-O
- S-Prot-Fr
- S-B2Miglo
- S-HIVAgAb, S-HBsAg, HBcAb, S-HCVAb, S-CMVAb
Luuydinnäytteet
- harkinnan mukaan
Sytogeneettiset tutkimukset
- FISH menetelmällä veren lymfosyyteistä
- 13q-deleetio
- 11q-deleetio
- 17p-deleetio ja
- trisomia 12.
- TP53 mutaatiostatuksen tutkimista suositellaan sellaisilta potilailta, joilla ei todeta 17p-deleetiolta.
- Sytogenetiikka tulee tutkia myös uusiutuneen taudin hoidon alkaessa, koska 17p-deleetio / TP53-mutaatio ilmaantuu usein taudin edetessä.
- IGHV- mutaatiostatus (B/Bm-IgSHM-D) tutkitaan kertaalleen (tulos muuttumaton) aktiivisen hoidon piirissä olevilta potilailta.
Kuvantamistutkimukset
- Ennen hoitoa kaikista potilaista otetaan thorax-röntgen ja vatsan UÄ.
- Vain erityistilanteissa tutkitaan vartalon TT-kuvaus, josta aiheutuva säderasitus on merkittävä.
Imusolmukkeen histologinen tutkimus
- Jos herää epäily taudin transformaatiosta, tulee suurentunut imusolmuke poistaa tuorenäytteeksi patologille. Edustavan imusolmukkeen valinnassa voi PET-TT olla hyödyllinen.
KLL:n seurantaohje
- Kirjaudu tai rekisteröidy kommentoidaksesi
KLL:n seurantaohje
Oireetonta ja pienimassaista KLL:aa seurataan ilman lääkehoitoja jopa vuosien ajan ja osalla tauti ei koskaan etene oireiseksi. Oireetonta tai vähäoireista tautia ei hoideta, koska hoidoista aiheutuisi enemmän haittaa kuin itse taudista. Liian varhaisen hoidonaloituksen ei ole todettu parantavan potilaan kokonaisennustetta. Useimmilla tauti kuitenkin etenee hitaasti ja on hyödyllistä seurata taudin etenemistä harvakseltaan mahdollisten hoidon aloituksen tarpeen ja komplikaatioiden varalta. Annettujen hoitojen jälkeen päästään usein vuosia kestävään oireettomaan vaiheeseen, jolloin seuranta tapahtuu samaan tapaan. Seuranta voi tapahtua avoterveydenhuollossa esimerkiksi kerran tai kaksi vuodessa.
Kontrollikäyntien yhteydessä
- kysytään yleisoireilusta (yleiskunnon muutokset, tahaton laihtuminen, infektioiden esiintyminen, yöhikoilu, kuumeilu)
- tehdään kliininen tutkimus: palpoidaan imusolmukealueet eli kaula, kainalot, nivusalueet ja arvioidaan pernan ja maksan koko
- tarkistetaan täydellinen verenkuva, TVK
Hoidon aloituksen aiheita ovat
- KLL:aan liittyvä anemisoituminen (Hb alle 110 g/l tai lasku 20g/l) tai trombosytopenia (B-Trom alle 100 x 10E9/l)
- erittäin kookas tai nopeasti kasvava perna, joka aiheuttaa oireita
- erittäin runsaat, oireita aiheuttavat tai nopeasti lisääntyvät imusolmukesuurentumat
- nopeasti lisääntyvä lymfosytoosi ( B-Lymf kaksinkertaistuu 6 kk:ssa tai nousu +50% 2 kk:ssa lähtötason ollessa yli 30). Pelkästään korkea, hitaasti nouseva/tasainen lymfosyyttitaso ei ole hoidon aihe, ellei siihen liity hyperviskositeettia
- kortikosteroidille reagoimaton autoimmuunihemolyyttinen anemia (AIHA) tai immunologinen trombosytopenia (ITP)
- voimakas yleisoireisuus
- tahaton laihtuminen (vähintään 10% painosta)
- selittämätön, voimakas ja toimintakykyä heikentävä väsymys
- kuumeilu yli 38C ilman infektiota vähintään 2 viikon ajan
- voimakas yöhikoilu vähintään 1 kk:n ajan ilman infektiota
KLL:aan liittyy lisääntynyt infektioherkkyys, jonka vuoksi kaikille suositellaan vuosittaista influenssarokotetta ja yleensä myös pneumokokkirokotetta.
Autoimmuunisytopeniat (AIHA ja ITP) liittyvät yleisesti KLL:aan. Yleensä ne reagoivat hyvin kortisonihoidolle.
Mikäli potilas on oireinen tai tauti osoittaa nopean etenemisen merkkejä tai siihen liittyy hankalia komplikaatioita, kannattaa hematologia konsultoida.
Valikon tekstit korjattu
KML
- Kirjaudu tai rekisteröidy kommentoidaksesi
KML
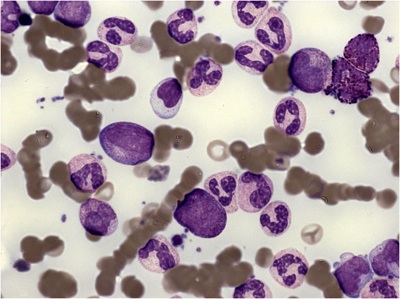 Krooninen myelooinen leukemia (KML) on krooninen, pahanlaatuinen hematopoieettisten kantasolujen sairaus. Uusia KML-potilaita todetaan Suomessa 45- 50/vuosi. Valtaosalla KML-potilaista tauti todetaan kroonisessa vaiheessa sattumalöydöksenä poikkeavan verenkuvan jatkoselvittelyissä.
Krooninen myelooinen leukemia (KML) on krooninen, pahanlaatuinen hematopoieettisten kantasolujen sairaus. Uusia KML-potilaita todetaan Suomessa 45- 50/vuosi. Valtaosalla KML-potilaista tauti todetaan kroonisessa vaiheessa sattumalöydöksenä poikkeavan verenkuvan jatkoselvittelyissä.
KML:n diagnoosi perustuu luuytimen tai veren soluista tehtävään BCR/ABL1- fuusiogeenin osoittamiseen. Klassisesti sen merkkinä todetaan kromosomitutkimuksessa (G-raitatutkimus) Philadelphia-kromosomi syntyneenä joko vakiotranslokaation t(9;22)(q34;q11) tai varianttitranslokaation pohjalta (n. 10 % potilaista).
BCR-ABL1 -fuusiogeeni on osoitettavissa PCR-tekniikan avulla myös niillä potilailla, joilla ei saada varmuutta Philadelphia-kromosomista (n. 2-3 % KML-potilaista).
KML-taudin hoidon perusta on tyrosiinikinaasin estäjähoito.
| Kirjoittajat: | Satu Mustjoki, Perttu Koskenvesa, Kimmo Porkka, Veli Kairisto, Hanna Rajala |
| © Suomen Hematologiyhdistys | Suomen Leukemiaryhmä |
Diagnoosi
- Kirjaudu tai rekisteröidy kommentoidaksesi
Diagnoosi
KML-epäily herää yleensä verikokeissa todetun leukosytoosin perusteella. Lisäksi voi olla trombosytoosia ja anemiaa. Perusterveydenhuollossa leukosytoosin luonnetta on syytä tarkentaa tekemällä valkosolujen erittelylaskenta, jossa tyypillisesti nähdään neutrofiilivaltainen leukosytoosi ja vasemmalle siirtymä granulopoieesin varhaismuotoina. Jatkotutkimukset on järkevintä tehdä hematologian yksikössä. Yleensä riittää pikaisesti tehty polikliininen selvittely, mutta KML:n takia oireinen potilas tai korkean leukosyyttitason takia erityistoimia tarvitseva potilas on syytä lähettää päivystyksellisesti sairaalaan. Diagnoosi voidaan nopeimmin tehdä PCR-tutkimuksen avulla verinäytteestä ja diagnoosin tarkentavat luuydinnäytteet sitten suunnitella halutussa laajuudessa.
Verikokeet
- TVK, CRP, ALAT, Afos, Bil, LD, Krea, K Na, Ca-alb-K, Mg, Pi, uraatti, CK, veriryhmä, TT, S-CMVAb, S-IgA, S-IgM, S-IgG
- Veren kvantitatiivinen BCR-ABL1 RQ-PCR. Käytetyt näyteputket ja toivotut verimäärät vaihtelevat laboratoriokohtaisesti.
- HUSLAB B-KML-Qr (21252) , TYKSLAB B-CMLPCR (12138) , NordLab B-BCR-Qr (4894), Fimlab B-BCR-QR(4894)
-
Kvantitatiiviset KML- seurantaan tarkoitetut PCR- testit mittaavat p210 (major, b2a2 (e13a2) ja b3a2 (e14a2)) transkriptejä. Harvinaisia transkriptimuotoja (b1a2 tai b3a3) epäiltäessä on syytä pyytää fuusiogeeniseulontaa. Ph+ALL:ssa esiintyvä p190 (e1a2)- transkripti todetaan ja kvantitatiivisesti seurataan B-BCR-Qr- tutkimuksella (4894).
- sokeri- ja kolesterolitasapaino erityisesti, jos suunnitteilla nilotinibi- tai ponatinibihoito
- TKE- lääkitystä edeltävästi suositellaan tarkistamaan HBV- status
Luuydinnäytteet
- Välttämättömät tutkimukset
- Luuytimen morfologia aspiraationäytteestä (MGG-värjäys)
- Luuytimen hematologinen kromosomitutkimus (Bm-KromHem), joskus pyydettävä pikatutkimuksena (valmistuu 2-3 vuorokaudessa)
- Mahdolliset lisätutkimukset ja näytteet
- Virtaussytometrinen pintamerkkitutkimus käytännössä otetaan blastikriisivaiheen potilaasta.
- FISH-tutkimus, tehdään usein rutiinisti kromosomilöydöksen jälkeen fuusiogeenin varmistamiseksi ja erityisesti, mikäli G-raita on jäänyt negatiiviseksi eikä kvantitatiivinen b-bcr-qr- tutkimuskaan ole informatiivinen.
- Fuusiogeenitutkimus on mahdollista tehdä luuydinnäytteestä, mutta seurantavaiheessa on syytä käyttää verestä tehtävää määritystä.
- Luuydinbiopsia, mikäli aspiraatin saaminen on ongelmallista tai epäily muusta hematologisesta sairaudesta
- FHRB- näytteet potilaan suostumuksella
Muut tutkimukset
- Anamneesi: muut sairaudet ja lääkitykset,luontaistuotteiden käyttö, tupakointi, alkoholinkäyttö, allergiat, työperäiset altistukset, sukuhistoria
- Ylävatsan UÄ, thorax-rtg
- EKG
- pituus, paino, verenpaine. Pernan palpointi on tärkeä tehdä ja tulos kirjata ennen mitään KML- hoitoa.
- sydämen uä, jos tiedossa sydämen vajaatoiminta tai muu selkeä sydänsairaus
Diagnoosi, B-lausunto ja rekisteröinti
- KML- diagnoosi perustuu BCR/ABL1- fuusiogeenin osoittamiseen ja/tai Ph-kromosomin osoitukseen luuytimen tai veren kromosomitutkimuksessa. Kvantitatiivisen PCR-seurannan mahdollisuuden varmistamiseksi PCR-näyte on syytä ottaa diagnoosivaiheessa.
- B-lausunto tulisi tehdä heti diagnoosin varmistuttua (mainittava Ph/BCR-ABL1-positiivisuus). Dg C92.1, koodi 117.
- B-lausunnolla tulee anoa samalla ensilinjaan valittua lääkettä eli samalla joko imatinibi-, nilotinibi- tai bosutinibihakemus. Koodi 189 imatinibi, 152 nilotinibi ja 170 bosutinibi.
- Potilas tulisi rekisteröidä Suomen hematologiseen rekisteriin heti diagnoosin varmistuttua. Rekisteröitymisen yhteydessä on hyvä tallentaa rekisteriin ne muutamat verikoelöydökset, joiden avulla lasketaan automaattisesti mm. Sokal- ja Euro/Hasford-pisteytykset kroonisen vaiheen potilailla. 2020 julkaistun ELN-suosituksen mukaisesti keskeisin riskiluokitus on ELTS, jonka laskuriin linkki luokitteluosiosta.
Luokitus
- Kirjaudu tai rekisteröidy kommentoidaksesi
Luokitus
Siirry riskiluokituksiin ja taudin vaiheet osioon sivupalkin linkeistä.
Riskiluokitus
- Kirjaudu tai rekisteröidy kommentoidaksesi
Riskiluokitus
- Tärkein riskiluokitus 2020 ELN-suosituksen mukaan on ELTS.
- Perustuu modernisti TKE-hoidettuihin potilaisiin toisin kuin aiemmat, joiden pohjalla TKE-aikakautta edeltäneillä tavoilla hoidetut.
- ELN 2020- suosituksessa korkean riskin ryhmään kuuluvat myös kromosomitutkimuksessa Ph-kromosomin lisäksi tiettyjä lisämuutoksia omaavat (+8, toinen Ph-kromosomi (+Ph), i(17q), +19, −7/7q-, 11q23 tai 3q26.2 muutokset ja kompleksi karyotyyppi.
- Huonompi todennäköisyys saavuttaa tavoitteen mukainen hoitovaste ja suurempi progression riski.
The EUTOS long-term survival (ELTS) score
- antaa prospektiivisen arvion ensilinjan imatinibihoidon tehosta ja elossaolon todennäköisyydestä.
- Korkean riskin ryhmään päätyy pienempi osuus potilaista, mutta heidän ennusteensa on selvästi huonompi, joten seuranta oltava tarkkaa ja lääkehoidolliset valinnat yksilöllisiä.
-
syytä laskea ja kirjata sairauskertomukseen diagnoosivaiheessa arvoilla, jotka mitattu ennen mitään KML-hoitoa.
- pisteytykseen vaadittavat tiedot
- Ikä (vuosia)
- Pernan koko (cm kylkikaaresta alaspäin palpoiden)
- B-blastit (%)
- B-tromb (10e9/L)
- Pisteet lasketaan syöttämällä edellämainitut arvot laskuriin.
- Riskiluokat
- pisteet ≤1.5680, pieni riski
- pisteet 1.5681- 2.2185, keskiriski
- pisteet > 2.2185, suuri riski
Sokal-pisteytys
- kehitetty ennen tyrosiinikinaasinestäjien (TKI) aikakautta, mutta ennustaa hoitovastetta myös TKI:lle
- tarpeellinen tieto, jos ensilinjan imatinibi-hoito joudutaan vaihtamaan 2. polven TKI:n huonon hoitovasteen vuoksi (Hammersmith-pisteytys)
- Pisteytykseen vaadittavat tiedot
- Ikä (vuosia)
- Pernan koko (cm kylkikaaresta alaspäin palpoiden)
- B-tromb (10e9/L)
- Myeloblastien (B-blastit) osuus veressä (%)
- Pisteet lasketaan syöttämällä edellämainitut arvot laskuriin. HUOM! Ole tarkkana solu-osuuksien kohdalla!
- Pisteet tulevat automaattisesti laskettua myös kun potilas tallennetaan SHR:in.
- Riskiluokat
- pisteet <0.8, pieni riski
- pisteet 0.8-1.2, keskiriski
- pisteet >1.2, suuri riski
Hasford (Euro)-pisteytys
- Ennustaa hoitovastetta Sokal-pisteytyksen tavoin
- Pisteytykseen vaadittavat tiedot
- Ikä (vuosia)
- Pernan koko (cm kylkikaaresta alaspäin palpoiden)
- B-blastit (%)
- B-eosinofiilit (%)
- B-basofiilit (%)
- B-tromb (10e9/L)
- Pisteet lasketaan syöttämällä edellämainitut arvot laskuriin. HUOM! Ole tarkkana solu-osuuksien kohdalla!
- Pisteet tulevat automaattisesti laskettua myös kun potilas tallennetaan SHR:in.
- Riskiluokat
- pisteet <780, pieni riski
- pisteet 780-1480, keskiriski
- pisteet > 1480, suuri riski
The EUTOS score
- Lasketaan samalla todennäköisyyttä OLLA SAAVUTTAMATTA tavoitevastetta CCyR 18kk hoidolla imatinibilla.
- Pisteytykseen vaadittavat tiedot
- Pernan koko (cm kylkikaaresta alaspäin palpoiden)
- B-basofiilit (%)
- Pisteet lasketaan syöttämällä edellämainitut arvot laskuriin.
- Riskiluokat
- pisteet ≤ 87, pieni riski
- pisteet > 87, suuri riski
Taudin vaiheet
Taudin vaiheet
Luokitus perustuu WHO2008-määritelmään. Hoidon kannalta keskeistä on kuitenkin erottelu kroonisen vaiheen ja edenneen taudin välillä.
Krooninen vaihe
- blastien määrä veressä ja luuytimessä <10%
- (ELN määritelmä, joka yleensä käytössä kroonisen vaiheen lääketutkimuksissa)
-
< 15% blasts in peripheral blood and bone marrow
-
< 30% blasts plus promyelocytes in peripheral blood and bone marrow
-
< 20% basophils in the peripheral blood
-
≥ 100 x 109/L (≥ 100,000/mm3) platelets
-
Kiihtynyt vaihe eli akseleraatiovaihe (vähintään yksi seuraavista)
- blastien määrä veressä ja/tai luuytimessä 10-19% (ELN 15-19%)
- veren basofilia ≥20%
- hoitoon liittymätön B-tromb <100 x10e9/L
- hoitoon reagoimaton B-tromb >1000 x10e9/L
- hoitoresistentti leukosytoosi
- hoitoresistentti/lisääntyvä splenomegalia
- klonaalisen evoluution merkit kromosomitutkimuksessa (verrattuna diagnoosivaiheen löydökseen)
Blastikriisi (vähintään yksi seuraavista)
- blastien määrä veressä ja/tai luuytimessä >20%
- luuytimessä laajoja blastisaarekkeita (kristabiopsia)
- extramedullaarinen blastiproliferaatio (klorooma)
Seuranta ja vastearvio
- Kirjaudu tai rekisteröidy kommentoidaksesi
Seuranta ja vastearvio
KML:n seuranta ja tautitaakan arvio perustuu pääasiassa veren kvantitatiiviseen BCR-ABL1-transkriptin PCR-mittaukseen. Katso tarkemmin Diagnoosi- sivulta.
Seurantamuuttujat ja -tiheys
- Veren PCR- määritys 3 kk hoidon aloituksesta ja sen jälkeen 3kk välein ad 36kk
- 24 kk jälkeen PCR- määritys 6kk välein, jos MR4 (≤0,01%IS)- tasoinen vaste
-
Kvantitatiiviset KML- seurantaan tarkoitetut PCR- testit mittaavat p210 (major, e13a2 (b2a2) ja e14a2 (b3a2)) transkriptejä. Harvinaista transkriptimuotoa b3a3 voidaan seurata B-BCR-mR- tutkimuksella (HUSLAB) Transkriptin näkyminen vasta nested-herkistyksen jälkeen antaa vaikutelman syvemmästä vasteesta. p190 (e1a2)- transkripti kvantitatiivisesti seurataan B-BCR-Qr- tutkimuksella (4894).
- Luuytimen G-raitatutkimus on tarpeen ELN 2020 hoitosuosituksen mukaan vain, jos PCR- tuloksen perusteella hoitovasteen kehitys on puutteellista. JOillain potilailla on selkeä ristiriita sytogeneettisen ja molekyyligeneettisen vasteen välillä. Täysi sytogeneettinen vaste eli CCyR on vahva hyvän ennusteen merkki ja tukee ristiriitatilanteessa aiemman hoidon jatkamista. G-raitatutkimus on tärkeä tutkia epäiltäessä KML:n etenemistä ja klonaalista evoluutiota.
- Luuytimen morfologinen tutkimus (MGG-värjäys) on tarpeen seurannassa vain, jos todetaan huomattava vasteen heikkeneminen tai epäselvät soluarvomuutokset.
- Lokusspesifinen FISH-tutkimus ei ole rutiiniseurantatutkimus vaan käytetään ainoastaan erityistapauksissa
- Pienellä osalla potilaista kromosomitutkimuksen ja PCR- tutkimuksen tulokset ovat ristiriitaisia ja sytogeneettisen vasteen syvyyttä voi arvioida FISH- tutkimuksella, jolla päästään 0,1% herkkyyteen.
- Jos potilaalla on harvinainen BCR-ABL1-fuusiogeenivariantti, jota ei voida seurata kvantitatiivisella PCR-tutkimuksella. Tällöin FISH-tutkimusta voi käyttää jäännöstaudin seurannassa tarkentamaan sytogeneettisen vasteen syvyyttä ≈ 0.1% tasolle
Verikokeet
- Perusverenkuva ja solujen erittelylaskenta
- 1-2vk välein ad 3kk
- 1-2 krt/kk ad 6kk
- 1 krt/kk ad 12kk
- 12kk jälkeen 3-6 kk välein
- Muut verikokeet
- Krea, alat, afos, bil
- 2-4x/kk välillä 0- 6kk
- x1/kk välillä 6- 12kk
- 12kk jälkeen 3- 6kk välein
- K, Pi, Mg, Ca-albK, CK, amyl, Bil-Kj
- tarpeen mukaan riippuen käytetystä lääkityksestä ja sivuoireista
- elektrolyytit 2-3x välillä 0-3kk
- 12kk hoidon aloituksesta: IgG, IgM, IgA
- Krea, alat, afos, bil
- Diagnostisten tutkimusten jälkeen, ennen tyrosiinikinaasin estäjähoidon aloitusta, mikäli leuk>50 ja/tai trom>500 on verenkuvaseuranta myös tarpeen n. 2vk välein
Muut tutkimukset
- EKG, kun hoitoannos ollut käytössä 2vkoa QT-ajan kontrolloimiseksi
- Pernan koon seuranta palpoiden käyntien yhteydessä
- Paino, verenpaine, sokeri- ja kolesterolitasapaino vuosittain riippuen käytetystä lääkityksestä
- Thx-rtg, EKG ja sydämen uä-tutkimus epäiltäessä hoidon vaikutusta sydän-keuhko- tilanteeseen
Vastearvio
- vaste arvioidaan ELN-kriteerien mukaisesti ja luokitellaan/kirjataan kuten Taudin status-kohdassa on kuvattu.
BCR-ABL1-mutaatiotutkimus
- Kirjaudu tai rekisteröidy kommentoidaksesi
BCR-ABL1-mutaatiotutkimus
BCR-ABL1-geenin mutaatiotutkimus on tärkeä selvitettäessä relapsin syytä tai huonoa hoitovastetta KML- ja Ph+ALL-taudeissa. Mutaatiohaussa löytyvät yleensä yleisimmät resistenssiä aiheuttavat mutaatiot. Mikäli tautitaakka on pieni (<1%), sekvensointimenetelmän herkkyys ei yleensä ole riittävä mutaatioiden toteamiseen (poikkeuksena kvantitatiivinen T315I-PCR, ks. alla)
Tutkimusaiheet
- Vasteen menetys tai huono hoitovaste tyrosiinikinaasiestäjähoidon aikana
Tutkimusmenetelmät
- Mutaatiohaku BCR-ABL1-geenin kinaasialueen sekvensointimenetelmällä
- T315I mutaation kvantitatiivinen osoitus PCR-tutkimuksen avulla
- NGS- ja ddPCR- menetelmillä löydetään mutaatioita vähäisemmästä tautimäärästä. Todettujen mutaatioiden kliininen merkitys ja lääkevaihdon tarve niiden pohjalta on edelleen tutkimuksen kohteena. 9/2021 analytiikka on käytettävissä SCANALL- tutkimuksen kautta. (HUS Syöpäkeskus Helena Hohtari, Perttu Koskenvesa)
Tulkinta
- T315I- mutaatio aiheuttaa resistenssin kaikille 1. ja 2.polven TKE- lääkkeille. Ponatinibi on tarjoutuva vaihtoehto.
- Ponatinibille resistenssiä aiheuttavia mutaatioita ei yksittäisinä ole, mutta kombinoidut mutaatiot voivat olla ongelma.
- T315I- mutaation ilmaantuminen on ennusteen kannalta huono asia ja mahdollisuudet kantasolujensiirtoon on syytä selvittää viipymättä. Ponatinibilla on saatu pitkäaikaisiakin vasteita, mutta riskinä on vasteen menetys, jolloin ennusteen kannalta toimivaa lääkehoitoa ei juuri ole tarjolla.
- askiminibi tarjolla managed access programin kautta (9/2021 Novartis)
- ABL-estäjä, jonka sitoutumistapa fuusioproteiiniin eroaa käytössä olevien TKE-lääkkeiden hyödyntämästä (myristoyylitasku vs ATP-sitoutumispaikka)
- ABL-spesifisyys voi vähentää muiden off target- kinaasikohteiden kautta selittyviä haittoja
- ABL-estäjä, jonka sitoutumistapa fuusioproteiiniin eroaa käytössä olevien TKE-lääkkeiden hyödyntämästä (myristoyylitasku vs ATP-sitoutumispaikka)
- hydroksiurea ja muut solunsalpaajat tarjoavat lähinnä keinoja leukosytoosin hillintään.
- Alfainterferonilla voidaan yksittäistapauksissa saada huomattavan pitkäkestoisiakin vasteita eli mutaation ilmaantuminen ei yksinään muuta KML:n biologista luonnetta.
- 9/2021 käytettävissä vai pegyloituja muotoja eikä niitäkään ole mahdollista määrätä reseptillä
- On mahdollista, että käyttöön tulee tutkimuslääkkeitä, joilla voi olla tehoa T315I-mutaatioon
- askiminibi tarjolla managed access programin kautta (9/2021 Novartis)
- Dasatinibiresistenssiin liittyvät mutaatiot: T315I, T315A, F317L/V/I/C, V299L. F317L/I-mutaatio on usein herkkä nilotinibille
- Nilotinibiresistenssiin liittyvät mutaatiot: T315I, Y253F/H, E255K/V, F359V/I/C
- Bosutinibiresistenssiin liittyvät mutaatiot: T315I, V299L, E255K
- Imatinibiresistenssiin liittyvät mutaatiot: kts. yst oheinen tiedosto.
Kirjallisuutta
- Impact of BCR-ABL mutations on patients with chronic myeloid leukemia. Hochhaus A, La Rosée P, Müller MC, Ernst T, Cross NC. Cell Cycle. 2011 Jan 15;10(2):250-60
- Mutations in the BCR-ABL1 Kinase Domain and Elsewhere in Chronic Myeloid Leukemia. Soverini S, de Benedettis C, Mancini M, Martinelli G. ClinLymphMyelLeuk. 2015; 15(S1):120-8
| Liite | Koko |
|---|---|
| 214.44 KB |
ELN-kriteerit
ELN-kriteerit
Vastearviokriteerit ensilinjan hoidolle. Tulee noudattaa myös 2. linjan hoidossa.
Vuonna 2020 päivitetty European LeukemiaNet:in (ELN) seuranta- ja vastearvio-ohjetaulukko. Kriteerejä käytetään arvioitaessa ensilinjan hoitoa. Akseleraatiovaiheen potilaille ei ole erillistä vastearviotaulukkoa ja vähintään samat kriteerit tulee täyttyä kuin kroonisen vaiheen taudissa.
|
Ajankohta |
Optimaalinen hoitovaste |
Varoitusmerkki |
Epäonnistunut hoitovaste |
|
|
Diagnoosi |
- |
Korkea ELTS- riskiryhmä Korkean riskin sytogen. poikkeavuus Ph+ soluissa +8, +Ph, i(17q), +19, −7/7q-, 11q23, tai 3q26.2- muutokset tai kompleksi karyotyyppi |
- |
|
|
3 kk |
BCR-ABL ≤10%(IS) |
BCR-ABL >10%(IS) |
Ei täyttä hematologista vastetta. BCR-ABL >10%(IS) uusittuna 1-3 kk kuluessa |
|
|
6 kk |
BCR-ABL ≤1% |
BCR-ABL >1-10%(IS) |
BCR-ABL >10%(IS) |
|
|
12 kk |
BCR-ABL ≤ 0.1%(IS) |
BCR-ABL >0.1-1%(IS) |
BCR-ABL >1%(IS) |
|
|
|
||||
|
Milloin tahansa |
MMR (MR3, ≤0,1%) tai parempi |
BCR-ABL >0.1-1%(IS), MMR-vasteen menetys
|
>1%(IS), resistenssimutaatiot, korkean riskin kromosomimuutosten ilmaantuminen
|
|
Viitteet:
Hochhaus et al European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia, Leukemia (2020) 34:966–984
| Liite | Koko |
|---|---|
| 663.23 KB |
Hammersmith-pisteytys
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hammersmith-pisteytys
Hammersmith-pisteytystä käytetään ennustamaan 2. polven tyrosiinikinaasiestäjällä saavutettavaa vastetta, kun joudutaan siirtymään toisen polven tyrosiinikinaasinestäjiin imatinibihoidon tehottomuuden vuoksi.
Taustaa
- Potilaat, joilla on primaarinen tai hankittu resistenssi imatinibille, saavuttavat täydellisen sytogeneettisen vasteen toisen polven tyrosiinikinaasinestäjille vain noin 50% tapauksista
- Noin 15% menettää sen kahden vuoden seurannassa
- Hammersmith-pisteytys on kehitetty löytämään mahdolliset kantasolusiirtoa tarvitsevat potilaat imatinibi-hoidon epäonnistuttua puutteellisen vasteen vuoksi
Pisteiden lasku ja tulkinta
|
Muuttujat |
Vaste |
Pisteet |
|
Paras sytogeneettinen vaste imatinibihoidon aikana |
CCyR 1-94% Ph+ >95% Ph+ |
0 1 3 |
|
Sokal-pisteet
|
Matala Keski-/korkea riski |
0 0.5 |
|
Neutropenia *
|
Ei Kyllä |
0 1 |
- *Neutropenialla tarkoitetaan imatinibihoidon aikana todettua toistuvaa gradus III-IV neutropeniaa (B-neutrofiilit <1x10E9 /L), joka on vaatinut imatinibin annoksen laskua alle 400 mg/vrk huolimatta kasvutekijähoidosta
- Pisteet <1.5 ennustavat hyvää vastetta 2. polven tyrosiinikinaasinestäjälle
- Pisteet ≥2.5 ennustavat huonoa vastetta, jolloin kantasolusiirtoa tulisi harkita (jos EBMT-pisteet suotuisat)
- Pisteet ≥1.5 mutta <2.5 ovat keskiriskin aluetta
Lääkepitoisuus
- Kirjaudu tai rekisteröidy kommentoidaksesi
Lääkepitoisuus
Tällä hetkellä (5/2018) vaikutelma on, että TKE- lääkkeiden pitoisuusmittauksilla ei ole käytännön merkitystä kliinisessä työssä. Pitoisuusmittauksia tehdään lääketutkimusten puitteissa ja HY Kliinisen farmakologian laitoksella on tutkittu ainakin imatinibin pitoisuuteen vaikuttavia asioita. Jos tarvetta pitoisuusmittaukselle tulee, voinee imatinibin ja nilotinibin osalta olla yhteydessä Novartis Finland Oy:n henkilöstöön mahdollisen näytteen lähettämismahdollisuuden suhteen.
Indikaatiot
- puutteellinen hoitovaste ja mietitään lääkkeen vaihtamista tai annoksen suurentamista
- poikkeavan voimakkaat sivuvaikutukset hyvän hoitovasteen omaavalla potilaalla
- lääkeinteraktioepäily
Toteutus
- Suomessa laboratoriomäärityksiä ei tällä hetkellä ole rutiinisti tarjolla.
- Suomesta on näytteitä lähetetty Ranskaan Bordeauxin laboratorioon. Aiemmin Novartis Finland Oy:ltä oli mahdollista saada näytteenottopakkaus, jossa oli myös tarkemmat näytteenotto- ja lähetysohjeet
Tulkinta
- Imatinibilla 1000ng/ml on pidetty raja-arvona, jonka on osoitettu korreloivan hyvän hoitovasteen saavuttamiseen (Picard et al Blood 2007)
- tuloksen mukana Bordeaux:stä tuli lausunto, jota varten potilaasta oli hyvä antaa mukaan tietoja hoitovasteesta, sivuvaikutuksista ja muista käytössä olevista lääkkeistä
- Matalan pitoisuuden tilanteessa vasteen paraneminen on varsin todennäköistä imatinibin annosnostolla. Haittavaikutusten lisääntymisen riski on olemassa mutta geneerisen imatinibin avulla annosnoston kustannusvaikutus on kohtuullinen ja tapauskohtaisesti harkittavissa, jos haittavaikutuksia normaaliannoksella on ollut vähän. Tätä ajattelua voidaan soveltaa myös ilman pitoisuusmittausta. Puutteellisen vasteen ja vähäisten haittojen tilanteessa voidaan tehdä oletus, että pitoisuus on KML-mielessä matala ja annosnostolla teho voi parantua. Vastaavasti hyvän vasteen ja hankalien haittojen tilanteessa matalampi annostaso voi olla mielekäs.
Taudin status
Taudin status
Vastearvion yhteydessä pyritään tautitaakan arvio ja taudin tilanne luokittelemaan oheisen taulukon mukaisesti (SHR-muuttuja: status-cml). Taulukosta valitaan arviointihetkellä oleva paras vaste.
|
Koodi |
Lyhenne |
Nimike |
Määritelmä |
|
0 |
Dg |
Diagnoosivaihe |
|
| 1 |
CMR |
Täydellinen molekyyligeneettinen vaste |
|
| 2 |
MMR |
Merkittävä molekyyligeneettinen vaste |
|
|
3 |
CCyR |
Täydellinen sytogeneettinen vaste |
|
|
4 |
PCyR |
Osittainen sytogeneettinen vaste |
|
|
5 |
MinCyR |
Vähäinen sytogeneettinen vaste |
|
|
6 |
CHR |
Täydellinen hematologinen vaste |
|
|
7 |
SD |
Vakaa tauti |
|
|
8 |
Hypoplasia |
Dysplasia/hypoplasia |
|
|
9 |
RD |
Refraktaari tauti (RD) |
|
|
10 |
Relapsi (mol) |
Molekyyligeneettinen relapsi |
|
|
11 |
Relapsi (sytog)
|
Sytogeneettinen relapsi |
|
|
12 |
Relapsi (hemat) |
Hematologinen relapsi |
|
|
13 |
Relapsi (AP) |
Hematologinen relapsi; transformaatio kiihtyneeseen vaiheeseen |
|
|
14 |
Relapsi (BC) |
Hematologinen relapsi; transformaatio blastikriisiin |
|
|
90 |
Exitus-relapsi |
Exitus, relapsi |
|
|
91 |
Exitus-infektio |
Exitus, infektio |
|
|
92 |
Exitus-aplasia |
Exitus, aplasia |
|
|
93 |
Exitus-muu tauti |
Exitus toisesta taudista johtuen |
|
|
99 |
Exitus-tuntematon |
Exitus, syy tuntematon |
|
|
999 |
Poistunut seurannasta |
Seuranta loppunut (lost-to-follow-up) |
|
Hoito
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hoito
Ensilinjan hoito (krooninen vaihe)
- Ensilinjan lääkehoitotutkimus (jos käynnissä)
- Imatinibi 400 mg/vrk tai nilotinibi 300 mg x2 tai bosutinibi 400mg x1
- Imatinibi on ensisijainen, mutta 2.polven lääkkeitä syytä harkita korkean ELTS-riskin potilailla ja nuoremmilla potilailla, joilla TFR-kokeilu esimerkiksi raskaustoiveen takia nopeammalla aikataululla paikallaan.
- Mikäli leukosyyttiluku diagnoosivaiheessa oireettomalla henkilöllä on <100 ja trombosyyttiluku on <1000, voidaan B-lausunnon hyväksymistä odottaa ilman lääkitystä (verikokeet viikoittain). Mikäli leukosyyttiluku diagnoosivaiheessa on >100 ja/tai trombosyyttiluku on >1000 aloitetaan hydroksiurea 500 mg 2x2 annosta viikoittaisten verikokeiden perusteella muokaten (max. 3 g/vrk). Lisäksi oheen on syytä aloittaa allopurinoli 300 mg/vrk (muistettava lopettaa verenkuvan normaalistuttua) ja huolehdittava riittävästä nesteytyksestä (ad 3000ml/vrk). Mahdollinen hydroksiureahoito voidaan lopettaa TKE-hoidon alettua, kun leukosyyttiluku on <20-30 ja trombosyyttiluku <600.
- Hoitovasteen arvioimisen mahdollistamiseksi on tärkeää, että ensimmäisen kolmen hoitokuukauden aikana pyrittäisiin lääkitystä jatkamaan keskeytyksettä. Sytopenioita voidaan tarvittaessa hoitaa verituottein ja/tai granulosyyttikasvutekijällä tukien. Alkuvaiheen sytopeniat ovat usein suotuisan vasteen merkki ja johtuvat yleensä Ph- positiivisen hematopoieesin nopeasta väistymisestä ja terveen Ph- negatiivisen hematopoieesin hitaammasta toipumisesta. 3kk jälkeen jatkuvat sytopeniat sen sijaan voivat kertoa Ph- negatiivisen hematopoieesin vauriosta, jolloin hoitostrategia täytyy miettiä uudelleen ja luuydinnäytteen ja kromosomitutkimuksen uusiminen on herkästi tarpeen.
- Granulosyyttikasvutekijähoidon tarve: Hoidon turvallisen toteutumisen kannalta pyritään neutrofiilitaso pitämään yli tason 0,5x10E9/L. Osalla potilaista esiintyy hoitoa ongelmoittavaa sytopeniaa 2. ja 3. hoitokuukauden aikana. Tämä on todennäköisempää korkean riskin potilailla, joiden kohdalla hoidon jatkuminen keskeytyksettä on erityisen tärkeää. Kasvutekijätukea on syytä tarvittaessa hyödyntää keskeytyksettömän hoidon mahdollistamiseksi. Jos neutrofiilit laskevat <0.5x10E9/L, on esimmäisellä kerralla syytä antaa tilanteen toipua ilman tukitoimia vain tauottamalla lääkitys. Lääkitys voidaan aloittaa uudelleen, kun neutrofiilitaso on korjaantunut tasolle >1,0x10E9/L. Mikäli neutrofiilitaso uudelleen laskee välille 0,5-1,0x10E9/L, tulee harkittavaksi kasvutekijähoidon käynnistäminen painon mukaisin annoksin. 1-2 annosta viikossa yleensä riittää. TKE-lääke on syytä tauottaa, mikäli riittävää vastetta kasvutekijätuelle ei saada. Kasvutekijähoito on hyvä tauottaa viikkoa ennen mahdollista luuydinnäytteen ottoa. Kasvutekijätarve harvoin jatkuu enää 3-4 kk:n hoidon jälkeen, jos hoitovaste muuten on tavoitteiden mukainen.
Hoidon vaihtamisen aiheita:
- Intoleranssi (etenkin alkuvaiheessa kannattaa lääkitys testata uudelleen 1-2 vkon tauon jälkeen). Hoitovaihtoehto: toisen polven tyrosiinikinaasiestäjähoito (harvoin risti-intoleranssia)
- Hoidon epäonnistuminen (failure) ELN-kriteerien perusteella. Hoitovaihtoehdot: A. lääketutkimus, B. toisen polven tyrosiinikinaasiestäjähoito, C. Allogeeninen siirto potilailla, jotka edenneet AP/BC vaiheeseen tai potilailla, joilla T315I mutaaatio
Ensilinjan hoito (edenneet vaiheet - akseleraatiovaihe tai blastikriisi)
- Imatinibi 600-800 mg/vrk tai 2. polven lääke (bosutinibi, nilotinibi, dasatinibi), mikäli tiedetään, että potilaalla imatininiresistenssiä aiheuttava mutaatio
- Blastikriisi hoidetaan kuten akuutti Ph-positiivinen leukemia (TKI-+monisolunsalpaajahoito). Dasatinibi on monoterapiana tehokas induktiohoito, konsolidaatiohoitoon tulisi liittää monisolunsalpaajahoito. Allogeeninen kantasolujensiirto siirto on aiheellinen, mikäli tauti saadaan remissioon (vähintään täydellinen hematologinen vaste, mielellään CCyR tai MMR).
- Kiihtyneen vaiheen (AP) potilaat voivat saavuttaa ennusteellisesti riittävän vasteen (tavoite vähintään MMR) TKE- hoidolla, jolloin kantasolujensiirto on 2. linjan hoito
- Tiivis hoitovasteen seuranta ja tarvittaessa hoidon muuttaminen erityisen tärkeää
Toisen linjan hoito (resistenssi/relapsi)
- Lääkehoitotutkimus
- Nilotinibi 400 mg x2, dasatinibi 100 mg x1 tai bosutinibi 500 mg x1. Valinta tapahtuu potilaan taustasairauksien ja lääkkeiden sivuvaikutusprofiilin mukaan. Myös BCR-ABL1-mutaatiostatus voi vaikuttaa keskeisesti lääkkeen valintaan.
- Ponatinibi on ainoa tehoava lääke T315I- mutaatiotilanteessa.
- Ponatinibi aloitetaan 30- 45mgx1 ja annoslasku nopeasti tasolle 15mgx1, kun vaste osoitettu.
- Jos ensilinjan lääke on ollut 2.polven TKE, on harkittava 2.linjan hoitona ponatinibia, jos vaihdon syy on failure- tason resistenssi, vaikka mutaatiota ei todettaisikaan.
- Allogeeninen kantasolujensiirto harkiten, jos EBMT-pisteytys pieni (0-1) ja ikä alle 61-vuotta ja on todettu vähintään sytogeneettinen relapsi ensilinjan hoidolle
Myöhemmän linjan hoito
- Lääkehoitotutkimus
- Allogeenisen kantasolujensiirron harkinta, jos ei kunnollista vastetta kahdelle TKE:lle tavoitteen mukaisella annostuksella
- ponatinibi
- Imatinibilla aloittaneilla toinen 2. polven tyrosiinikinaasinestäjä on mahdollinen 3. linjassa, jos vaihdon syynä on intoleranssi
- alfainterferoni huonosti saatavilla
- hydroksiurea
- askiminibi MAP- ohjelman puitteissa. (tilanne 9/21)
| Liite | Koko |
|---|---|
| 663.23 KB | |
| 1.38 MB |
Kantasolujensiirto
Kantasolujensiirto
Ensilinjan hoidossa kroonisessa vaiheessa allogeeninen kantasolujensiirto tulee kyseeseen ainoastaan lapsilla. Aikuispotilailla kantasolujensiirron aiheita:
- 2. linjan hoidon epäonnistuminen (sytogeneettinen relapsi tai ei vähintään sytogeneettista vastetta ollenkaan)
- vaikea intoleranssi tyrosiinikinaasiestäjälääkityksille estäen ennusteen kannalta riittävän tehokkaan hoidon
- Edenneen vaiheen KML (blastikriisissä aina selvitettävä mahdollisuudet. Akseleraatiovaiheessa diagnosoiduilla, jos vajavainen vaste TKE-hoidolle tai akseleraatio hoidon läpi)
EBMT-riskiluokitus
EBMT-riskiluokitus
EBMT risk score definition
Risk factor Score point
Age of the patient
<20 years 0
20–40 years 1
>40 years 2
Disease stage
Early (= first chronic phase) 0
Intermediate (= accelerated phase) 1
Late (= other) 2
Time interval from diagnosis to transplant
<12 months 0
>12 months 1
Donor type
HLA-identical sibling donor 0
Unrelated donor 1
Donor recipient gender combination
All other 0
Donor female – male recipient 1
Erityiskysymyksiä
Erityiskysymyksiä
Muutamia TKE-hoitoon liittyviä erityiskysymyksiä käsitellään erillisissä teksteissä, joihin pääset sivupalkin linkeistä.
Geneerinen imatinibi
Geneerinen imatinibi
Oheinen tiedote on toimitettu nähtäväksi potilasverkoston FaceBookissa.
| Liite | Koko |
|---|---|
| 132.67 KB |
Geneerinen imatinibi
Geneerinen imatinibi
Oheinen tiedote laitettiin tiedoksi Potilasverkoston FaceBookin kautta
| Liite | Koko |
|---|---|
| 132.67 KB |
Lääkityksen tauotus ja annoksen lasku pitkäaikaishoidossa
Lääkityksen tauotus ja annoksen lasku pitkäaikaishoidossa
Nykytietämyksen mukaan tyrosiinikinaasinestäjälääkitys ei ole parantava hoitomuoto ja lääkityksen käyttöä tulee lähtökohtaisesti jatkaa koko elämän ajan. Lääkityksen tauotusta on kuitenkin tutkittu jo yli kymmenen vuoden ajan.
Ensimmäiset raportit ovat tulleet ranskalaisilta tutkijoilta. Ensimmäisessä tutkimusasetelmassa noin puolet potilaista, jotka olivat saaneet täydellisen molekyyligeneettisen vasteen (CMR) imatinibille ja olleet PCR-negatiivisia vähintään 2 vuoden ajan, kykenivät lopettamaan lääkkeen käytön ilman, että tauti ilmaantui näkyviin. STIM (Mahon et al, Lancet Oncol 2010). Näiden potilaiden osuus kaikista KML-potilaista on kuitenkin hyvin pieni (<10%).
Tauotuskokeilun edellytysten keventäminen ja korkeamman tautimäärän salliminen seurannassa ovat mahdollistaneet tauotuksen kokeilemisen laajemmalle potilasjoukolle. MR4- tasoisen vasteen saavuttaminen on mahdollista 40-60%:lle potilaista.
Suurimman tauotusta tutkineen kansainvälisen tutkimuksen tulokset on raportoitu toukokuussa 2018. EURO-SKI (Saussele et al, Lancet Oncol 2018). Raportin mukaan 24kk kohdalla MMR- vasteessa oli edelleen ilman lääkitystä 50% analysoiduista 755 tutkittavasta. Suomesta tutkimukseen osallistui 28 tutkittavaa. Lähinnä pohjoismaisista potilaista koostuneessa jatkoseurannassa tutkittavia seurattiin aina 6 vuoteen asti. Tärkein havainto oli, että 36kk kohdalla MR4-vasteessa säilyneistä vain 1/99 joutui aloittamaan lääkityksen uudelleen seurantavälillä 36-72kk. <MR4-vasteessa olleista lääkitys alkoi uudelleen 11/12 tutkittavalla. Tämän perusteella seurantaa on mahdollista harventaa 3v jälkeen 4kk välein tapahtuvaksi niillä, jotka ovat vähintään MR4-vasteessa. 6 vuoden jälkeen seurantaväli voi jo olla 6kk.
Tauotuskokeilun toteutus
Tauotuskokeilun suositeltavat edellytykset perustuen EURO-SKI- tutkimuksen tuloksiin:
- Kroonisen vaiheen KML, lääkevaihto sallittu intoleranssin takia
- Riittävä TKE-hoidon kesto (imatinibi 5-6v, nilotinibi 4v)
- Stabiili hoitovaste edeltävän vuoden ajan vähintään MR4 (3 mittausta)
Seuranta tauotuskokeilun aikana EURO-SKI- protokollan mukaisesti:
- TVK+ veren PCR x1/kk ensimmäisen kuuden kuukauden ajan
- TVK+ veren PCR 6vkon välein seuraavat 6kk
- TVK+ veren PCR 3kk välein 2. ja 3. vuosi ja sitten 3-4x/ vuosi pysyvästi
Lääkityksen uudelleenaloituksen raja on 0.1%(IS) eli tautimäärän säilyessä tämän tason alapuolella, voi lääketauko jatkua. Tiheämpi PCR-seuranta voi olla paikallaan 12kk jälkeen, jos tulostaso on lähellä 0.1% tasoa. Rajan ylittyessä aloitetaan aiempi lääkitys aiemmalla annostuksella uudelleen ja seuranta PCR:n osalta 3kk välein, kunnes MR4-vaste uudelleen saavutettu.
Tauotukseen on liittynyt lihas ja nivelkipuja, joiden ajatellaan johtuvan lääkityksen aikana estettynä olleinen tyrosiinikinaasikohteiden elpymisestä. Oireet ovat tyypillisesti ilmaantuneet muutaman viikon- 2kk aikana tauotuksesta ja kestäneet viikkoja ja jopa kuukausia. Verikokeissa ei ole nähty mitään immunologiseen aktiivisuuteen viittaavaa. Hoitona on käytetty NSAID ja lyhyitä kortisonikuureja tarvittaessa.
Lääkeannoksen alentaminen pitkäaikaishoidossa
Tauotuksen sijaan voidaan potilaan lääkeannosta portaittain alentaa ja seurata PCR-vasteen kehitystä 3kk välein 6kk ajan ennen seuraavaa muutosta. 3v hoidon jälkeen keskeistä on hyvän vasteen saavuttaneilla varmistaa lääkityksen pitkäaikaiskäytön siedettävyys. Hoidon tavoitteesta on hyvä sopia potilaan kanssa. MR4.5- MR5- tasoinen vaste on hyvä tavoite suunniteltaessa lääkityksen tauotuskokeilua. Mikäli sellaiseen ei tähdätä, on MR3- tasoisen vasteen säilyttäminen riittävä tavoite ja tähän voi 3v hoidon jälkeen riittää selvästi alkuperäistä annosta matalampi annostaso. Imatinibilla annosportaat luontevasti 400-300-200mg, nilotinibilla 300mgx2, 450mgx1, 300mgx1 ja dasatinibilla 100-70-50-20mg. Erityisesti dasatinibilla alennetuista annoksista on kertynyt kokemusta, koska monen potilaan kohdalla toistunut pleuranesteily on johtanut annoksen laskuun.
Brittitutkijat selvittelivät portaittaisen annoslaskun vaikutusta lääkityksen tauotuksen onnistumiseen 2017 julkaistussa tutkimuksessa. DESTINY (Clark et al, Lancet Haematol 2017). Onnistumistodennäköisyys ei ainakaan huonontunut. Pelkästään annoslaskun käytöstä pitkäaikaishoidossa ei ole raportteja.
Lisätietoja lääkityksen tauotukseen ja annoslaskuun liittyen voi kysellä HYKS/Syöpäkeskus/Hematologia ol Perttu Koskenvesa (perttu.koskenvesa@hus.fi)
| Liite | Koko |
|---|---|
| 348.01 KB | |
| 278.18 KB | |
| 1.58 MB |
Pleuraeffuusion hoito (dasatinibi)
- Kirjaudu tai rekisteröidy kommentoidaksesi
Pleuraeffuusion hoito (dasatinibi)
Dasatinibiin liittyy osalla potilaista nesteen kertymistä keuhkoihin. Ensilinjan hoidossa sivuvaikutuksen yleisyys on n. 10% luokkaa (Kantarjian et al, NEJM 2010) ja toisen linjan hoidossa 10-20% luokkaa. Riskitekijöinä pleuraeffusion saamiselle pidetään korkeaa ikää, aiempaa sydän- tai keuhkosairautta sekä mahdollisesti immunologisia sairauksia.
Ensisijaisena hoitona pleuraeffusion ilmaannuttua tulee lääke tauottaa. Tarvittaessa voidaan käyttää lyhytkestoista nesteenpoistolääkettä tai kortikosteroidia lisänä. Lääkkeen uudelleenaloituksessa tulee annostus usein olla aiempaa annostusta matalampi. Osalla potilaista sivuvaikutus voi uusia, mutta ei kaikilla.
Pleuraeffusion syntymisen tarkkaa mekanismia ei tunneta. Mahdollisesti se johtuu kuitenkin lääkkeen aiheuttamasta immunologisesta aktivoitumisesta. Tällä on osoitettu olevan yhteys myös paremman hoitovasteen syntyyn (Porkka et al, ASH2010).
Raskaus
Raskaus
KML-potilaiden keski-ikä diagnoosivaiheessa on yli 50 vuotta. Monen potilaan kohdalla lastenhankinta on kuitenkin edelleen ajankohtainen asia. On selvää, että potilaan sairaustilanteen on syytä olla vakaa ennen raskauden käynnistymistä. Sairastuminen krooniseen verisairauteen on psyykkisesti hyvin kuormittava asia ja sairauteen sopeutumiselle on annettava riittävästi aikaa.
Pääsääntö on, että naisen tulee huolehtia ehkäisystä tyrosiinikinaasin estäjähoidon aikana. Imatinibihoidon aikana käynnistyneistä raskauksista on olemassa kohtuullisesti tietoa, kun taas 2.polven lääkkeiden nilotinibin ja dasatinibin osalta ei käytännössä ollenkaan. Niiden osalta ongelmat tuskin ovat ainakaan vähäisempiä.
Imatinibihoidon aikana käynnistyneiden lapsen syntymään päättyneiden raskauksien perusteella on havaittu, että tiettyjen vaikeiden keskushermoston ja suoliston kehityshäiriöiden ilmaantuvuus on selvästi normaaliväestöä suurempaa. Vaikuttaakin siltä, että lääkkeen käyttö sikiönkehityksellisesti herkän 1. raskauskolmanneksen aikana on erityisen haitallista. Istukan kehityksen myötä voimakkaasti proteiineihin sitoutuva lääke ei enää läpäise istukkaa merkittävästi, jolloin lääkkeenkäyttö on tarvittaessa mahdollista myöhempien kolmanneksien aikana. Imatinibi erittyy rintamaitoon merkittävässä määrin, joten imetys ei lääkettä käytettäessä ole suositeltavaa.
Mikäli raskaus alkaa imatinibihoidon aikana, on lääkitys heti tauotettava. Raskauden jatkamisen osalta arvio on yksilöllinen, jossa potilaan sairaustilanne sekä sikiön imatinibille altistuma aika on huomioitava. Potilasta on informoitava lisääntyneestä vaikean kehityshäiriön riskistä, jos raskaus on havaittu selvästi viikon 7 jälkeen. Huomioitavaa on, että valtaosa imatinibihoidon aikana käynnistyneiden raskauksien seurauksena syntyneistä lapsista on ollut terveitä eikä heillä ole seurannassakaan havaittu lisäongelmia altistuksesta johtuen.
Toive raskaaksi tulemisesta vaatii TKI-lääkityksen keskeyttämistä. Edelleen vallitseva käsitys on, että valtaosalla taudin määrä alkaa lisääntyä heti lääkkeen loppuessa. MMR- vasteessa olevan potilaan kohdalla sytogeneettinen relapsi on hyvin todennäköinen <6kk aikana tauotuksesta. Korvaavan hoidon eli lähinnä interferonin käyttöä on syytä tämän takia harkita raskautta yritettäessä ja edelleen 1. kolmanneksen aikana. Ongelmana voi olla hoidon sieto. Potilailla, joiden lähtötilanne on vakaasti CMR-tasoinen, voi aikaa olla selvästi enemmän. TKI- lääkkeen tauotuksen jälkeen ja mahdollisen korvaavan lääkityksen aikana on suositeltavaa seurata verenkuvaa ja jäännöstautia kuukausittaisin määrityksin. Lääkkeenaloituskynnyksenä voidaan pitää IS-asteikolla tasoa 1,0%, joka vastaa suunnilleen G-raitatutkimuksen CCyR- tasoa. Aiemman kaltainen hoitovaste on säännönmukaisesti saavutettu lääkkeen uudelleenaloituksen jälkeen.
Siemennesteen talteenotosta on käytännössä voitu luopua imatinibia käyttävien lastenhankkimista edelleen harkitsevien miesten kohdalla. Siittiöiden määrässä ja liikkuvuudessa on havaittu heikentymistä, mutta estettä hedelmöittämiselle ei katsota olevan. 2.polven TKI- lääkkeisiin siirryttäessä on perusteltua edelleen tarjota siemennesteen tallentamismahdollisuutta tulevaa käyttöä ajatellen.
Tutkimukset
Tutkimukset
KML-perustutkimuksesta lisätietoa hematologisen tutkimusyksikön (HruH) kotisivuilla.
Ensilinjan tutkimukset
| Nimilyhenne | Rekrytointi | Seuranta | Tutkimuksen vastuuhenkilöt |
|---|---|---|---|
| BosuPeg (NCMLSG012) bosutinibi +/- ropegIFN | Alkanut | Perttu Koskenvesa, Satu Mustjoki | |
| B1871053 BFORE (bosutinibi) | Loppunut | Käynnissä | Perttu Koskenvesa, Satu Mustjoki, Kimmo Porkka |
Puutteellinen vaste, krooninen ja kiihtyneen vaiheen KML
| Nimilyhenne | Rekrytointi | Seuranta | Tutkimuksen vastuuhenkilö |
|---|---|---|---|
|
B1871040 bosutinibi |
Loppunut | jatkuu | Perttu Koskenvesa, Kimmo Porkka, Satu Mustjoki |
Muut tutkimukset
| Nimilyhenne | Rekrytointi | Seuranta | Tutkimuksen vastuuhenkilö |
|---|---|---|---|
| STOP | Päättynyt | Käynnissä | Kimmo Porkka |
| EURO-SKI (seuranta ad 6v) | Päättynyt | Käynnissä | Perttu Koskenvesa |
| DASTOP2 (2.lopetusyritys, dasatinibi 2v) | Avoinna | Satu Mustjoki | |
| NCMLSG013 (LTS) Laboratoriotutkimus tauotusta kokeileville (HRUHLAB2) | Avoinna | Anna Kreutzman |
BosuPeg
BosuPeg
Lyhyt yhteenveto tutkimussuunnitelmasta
Tutkimus:
A STUDY OF EFFICACY AND SAFETY OF LONG-ACTING LOW DOSE ROPEGINTERFERON IN PATIENTS WITH CHRONIC MYELOID LEUKEMIA TREATED WITH BOSUTINIB FROM DIAGNOSIS: A RANDOMIZED PROSPECTIVE TRIAL
Satunnaistettu, etenevä KML:n ensilinjan lääkehoitotutkimus: Lääkeaineen tehoa ja turvallisuutta mittaava tutkimus, jossa pieniannoksinen ropeginterferoni lisätään bosutinibihoitoon hiljattain diagnosoitua, kroonisessa vaiheessa olevaa kroonista myelooista leukemiaa sairastavilla potilailla.
Tutkimuksen koodi: BosuPeg
Tutkimuksen EudraCT- numero: 2018-001044-54
Krooninen myelooinen leukemia (KML) on pahanlaatuinen verisairaus, jonka syntymekanismi tunnetaan hyvin tarkkaan. Sairaudelle tyypillisenä muutoksena todetaan valtaosalla potilaista kromosomien 9 ja 22 välinen translokaatio, jonka seurauksena kromosomissa 9 oleva Bcr- geeni ja kromosomin 22 pitkässä haarassa oleva proto-onkogeeni Abl muodostavat sairaudelle tyypillisen fuusiogeenin. Tämä geeni tuottaa BCR-ABL- onkoproteiinia, jonka tyrosiinikinaasiaktiivisuus aiheuttaa näiden leukeemisten kantasolujen jakautumisaktiviteetin huomattavan kasvun.
Tämän perustavaa laatua olevan havainnon pohjalta onnistuttiin kehittämään kyseisen proteiinin vaikutuksen estäviä lääkkeitä, tyrosiinikinaasin estäjiä (TKE, imatinibi, nilotinibi, dasatinibi, bosutinibi, ponatinibi), jotka ovat mullistaneet KML:n hoidon. Käytännössä kaikille uusille kroonisen vaiheen potilaille käynnistetään TKE- hoito, joka on lähtökohtaisesti pysyvä. KML todetaan Suomessa 0.9/100 000 henkilöllä vuosittain eli uusia potilaita on vuosittain 40-45. Nykyisellään imatinibin rinnakkaisvalmisteiden myötä lääkityksen vuosikustannuksen haitari on 4000- 83 700 €/v.
TKE- hoidolla >90%:lla potilaista ennuste paranee niin, että KML:n ei katsota enää lyhentävän odotettavissa olevaa elinikää. Hoidon ansiosta osalla potilaista merkit KML-taudista katoavat täysin, eikä leukemiasoluja pystytä havaitsemaan edes herkissä laboratoriotutkimuksissa (PCR) tai tautimäärä on hyvin vähäinen ennusteen kannalta riittävään tasoon verrattuna. Tutkimukset ovat osoittaneet, että noin puolella näistä erinomaisen vasteen saavuttavista potilaista tautimäärä ei merkittävästi nouse, vaikka lääkehoito lopetettaisiin. Tauotuksen mahdollistavan vasteen saavuttaa KML-potilaista pitkällä aikavälillä 40-50%. Kaikista KML-potilaista siis 20-25% voisi 4-5 vuoden TKE-hoidon jälkeen mahdollisesti jatkaa seurannassa ilman lisähoitoa. Vastaavasti 75-80% potilaista tarvitsee TKE-lääkitystä pysyvästi altistuen ainakin elämänlaadullisesti harmillisille haittavaikutuksille. Tämä on motivoinut tutkijoita löytämään uusia hoitomuotoja, jotka voisivat mahdollisesti kasvattaa lääkkeen tauottamiseen kykenevien potilaiden osuutta. Ajatuksena on, että sairauden kontrollointiin ilman lääkitystä tarvitaan potilaan oman immuunijärjestelmän aktivoitumista. Sairautta ei TKE-hoidolla saada kokonaan hävitettyä mutta osalla potilaista on siis kehittynyt sairautta hillitsevä ominaisuus. Interferoni-alfa-hoitoa (IFN) käytettiin KML-taudin hoidossa ennen TKE-lääkityksen aikakautta. Myös IFN-lääkitys voi aikaansaada immuunijärjestelmän aktivoitumista vastaavalla tavalla ja osa potilaista on kyennyt vastaavasti lopettamaan IFN-lääkityksen ilman taudin uusiutumista. Tutkimukset ovat myös osoittaneet, että pegyloidun interferonialfan lääkkeen yhdistäminen imatinibi-, nilotinibi- tai dasatinibihoitoon edesauttaa paremman hoitovasteen saamisessa. Yhdistelmähoidon on todettu lisäävän niiden potilaiden määrää, joilla taudin tunnusmerkkejä ei enää havaita. Näin ollen on mahdollista, että interferonilääkkeen yhdistäminen TKE-lääkitykseen lisää niiden potilaiden määrää, joilta tulevaisuudessa pystyttäisiin lääkitys kokonaan lopettamaan.
Tutkimuksen tavoitteet
Tämän tutkimuksen tarkoituksena on yhdistää pitkävaikutteinen IFN-hoito (RoPEG-IFN, Besremi®) tehokkaaseen TKE-lääkitykseen (bosutinibi, Bosulif®). Yhdistelmähoitoa verrataan pelkkään bosutinibihoitoon. Hypoteesina on, että yhdistelmähoito lisää niiden potilaiden määrää, joilla päästän tauotuskokeiluun riittävän hyvän hoitovasteen ansiosta. Bosutinibi on osoitettu imatinibia tehokkaammaksi TKE-lääkkeeksi arvioitaessa hoitovasteen syvyyttä. Lisäksi tarkoituksena on tutkia niiden vaikutusta leukemiakantasoluihin ja immuunipuolustusjärjestelmään.
Tutkimuksen kulku
Tutkimukseen otetaan mukaan vastadiagnosoituja KML-potilaita, joilla ei ole aloitettu vielä muuta TKE-hoitoa KML:an. Mikäli tutkimuksen sisäänottokriteerit täyttyvät, tutkimuslääkitys bosutibilla aloitetaan 200 mg kerran vuorokaudessa annosteltuna. Annostusta nostetaan 1-2 viikon välein siedon mukaan tasolle 400mgx1. Siedettyä annostasoa käytetään 3kk, jonka jälkeen hoitoon lisätään pieniannoksinen (50ug/ 2 viikkoa) ropegyloitu IFNalfa2b puolelle tutkittavista. Yhdistelmähoitoa käytetään 21kk ajan, jonka jälkeen jatketaan pelkällä bosutinibilla. 48 kuukauden kokonaishoidon jälkeen edetään tauotuskokeiluun tutkittavilla, joiden hoitovaste on riittävä. Tutkimus kestää lääkkeettömän seurannan vaihe mukaan luettuna yhteensä yksittäisellä tutkittavalla maksimissaan 84kk ajan.

Tutkimuksen puitteissa lääkityksen aloittavien hoito tutkimuksen seitsemän vuoden keston aikana jakaantuu neljään osaan seuraavalla tavalla:
Osa 1
Bosutinibilääkitys aloitetaan kaikille päivittäisellä 200 mg annoksella. Suositeltavaa on ottaa lääkeannos ruokailun yhteydessä, mutta tablettien kanssa tulisi vähintään juoda iso lasillinen vettä. Ensimmäisenä lääkepäivänä otetaan verikoe lääkevaikutuksen arvioimiseksi ennen lääkkeenottoa ja 4 tuntia lääkkeenoton jälkeen. Lisäksi ennen lääkkeenottoa tutkittavien on täytettävä kaksi elämänlaatua mittaavaa kyselyä. Annosta nostetaan 1-3 viikon välein ensin tasolle 300mg päivässä ja edelleen 400mg päivässä, jos siedettävyyden kanssa ei ole ongelmia. Alkuvaiheessa verikoeseuranta ei oleellisesti eroa yleisen hoitosuosituksen mukaisesta. Lääkärintarkastus tehdään vastaanotolla yhden kuukauden hoidon jälkeen, muuten tuloksista ja hoito-ohjeista tiedotetaan puhelimitse. 3 kuukauden hoidon jälkeen hoitovastetta arvioidaan veri- ja luuydinnäyttein ja otetaan samassa yhteydessä tutkimusnäytteitä (90ml + 40ml), verikoe lääkevaikutuksen arvioimiseksi ennen lääkkeenottoa ja 4 tuntia lääkkeenoton jälkeen.
Osa 2
Kolmen kuukauden bosutinibilääkityksen jälkeen arvioidaan tutkittavan soveltuminen mahdolliseen yhdistelmähoitoon.Arpouttaminenvoidaan tehdä niille tutkittaville, jotka kykenevät käyttämään bosutinibia vähintään 300 mg päivä annoksella. Lisäksi varmistetaan vointi ja munuaisten- ja maksantoiminta yhdistelmähoidon mahdollisen suuremman kuormittavuuden takia. Tutkittavat, jotka eivät siedä bosutinibia kuin 200mg päiväannoksella eivät osallistu arpouttamiseen, mutta jatkavat tutkimuksessa bosutinibihoidolla edellyttäen, että hoitovaste kehittyy suotuisasti. Arpouttamisen jälkeen puolet tutkittavista jatkavat pelkällä bosutinibihoidolla ja puolet aloittaa bosutinibin rinnalle RoPegIFN-hoidon.
Ropeginterferonilääkitys (RoPegIFN) aloitetaan 50 mikrogramman annoksella kahden viikon välein. Lääkitys annetaan ihonalaisella pistoksella ja tutkittavat opetetaan itsenäisesti hoitamaan lääkkeen annostelu. Bosutinibilääkitys jatkuu tutkittavan 3 kuukauden alkuhoidon lopussa käyttämällä annoksella 300 mgx1 tai 400mgx1. Mikäli RoPegIFN aiheuttaa merkittäviä haittavaikutuksia, voidaan annostelua harventaa kolmen ja edelleen tarvittaessa neljän viikon välein tapahtuvaksi kerta-annoksen pysyessä ennallaan. Lääkärintarkastukset tehdään 4 kuukaudenja 6 kuukauden kohdalla ja sen jälkeen kolmen kuukauden välein 2 vuoteen asti. RoPegIFN on siis enimmillään käytössä 21 kuukautta. Aiempien tutkimustuloksien mukaan riittävä hyöty interferonista saavutetaan tässä ajassa. Lieväasteisten haittavaikutusten sietämiseen tutkittavia tullaan kannustamaan, koska yhdistelmähoidolla on aiemmin osoitettu pelkkää tablettihoitoa parempi hoitoteho.
Tutkimuksen keskeinen päätetapahtuma on tutkittavien saavuttama hoitovaste 12 kuukauden hoidolla. Tätä tutkitaan normaaliseurannan mukaisten veri- ja luuydinnäytteiden lisäksi ottamalla edellisten aikapisteiden kaltaisesti laboratoriotutkimusnäytteitä (90ml+ 40ml).
Osa 3
Välillä 24- 48 kuukautta kaikki tutkittavat käyttävät taas pelkkää bosutinibilääkitystä. Seuranta tapahtuu verikokein ja vastaanottokäyntejä on edelleen 3 kuukauden välein.
Osa 4
48 kuukauden hoidon jälkeen arvioidaan mahdollisuus lääkityksen tauotuskokeiluun. Tähän liittyy mahdollisesti myös luuydinnäyte tutkimustarkoitukseen. Hoitovasteen on täytynyt olla MR4-tasoinen edeltävän vuoden ajan. Tauotuskokeilun aikana verikokeita otetaan tautimäärän seuraamiseksi kuukausittain ensimmäisen 6 kuukauden ajan, sitten 6 viikon välein 12 kuukauteen asti ja tauon jatkuessa sen jälkeen 3 kuukauden välein. Mikäli hoitovaste ei mahdollista tauotuskokeilua 48 kuukauden kohdalla, jatkuu bosutinibihoito tutkimuksessa ja seuranta kuten osassa 3. Tauotuksen mahdollistava hoitovaste voi kehittyä hoidon jatkuessa myös myöhemmin. Tautimäärän tulee pysyä tasolla £0.1% IS, jotta lääketauko voi jatkua. Mikäli tautimäärä nousee tämän tason yläpuolelle, alkaa aiempi bosutinibilääkitys uudelleen ja seuranta tutkimuksessa jatkuu vasteen saavuttamisen seuraamiseksi kuten osassa 3. Seitsemän vuoden seuranta-ajan päättyessä ilman lääkehoitoa olevat tutkittavat jatkavat lääketaukoa normaaliseurannan puitteissa. Bosutinibia käyttävien tutkittavien lääkehoitoa anotaan korvattavaksi vallitsevien korvattavuusperusteiden mukaisesti.
| Liite | Koko |
|---|---|
| 313.62 KB | |
| 347.52 KB |
DASTOP2
DASTOP2
Lyhyt esittely:
Kyseessä on pohjoismainen tutkijalähtöinen monikeskustutkimus, jossa tutkitaan mahdollisuutta lopettaa tyrosiinikinaasin estäjähoito (TKE-hoito) erinomaisen vasteen saavuttaneilla kroonista myelooista leukemiaa sairastavilla potilailla. Aiemmissa kansainvälisissä tutkimuksissa on osoitettu, että n. 40% KML potilaista voi lopettaa TKE-lääkityksen ilman, että taudin määrä merkittävästi lisääntyy, mikäli he ovat käyttäneet riittävän pitkään TKE-lääkettä ja mikäli he ovat saavuttaneet erittäin hyvän hoitovasteen (Bcr/Abl- fuusiogeenin määrä alle 0.01%). Vastikään eurooppalaiseen EURO-SKI-tutkimukseen otettiin mukaan noin 800 potilasta, joilla oli erinomainen vaste TKE-hoidolle ja joista suurin osa oli hoidettu ensimmäisen polven TKE-lääkkeellä imatinibilla (Glivec). Kolme vuotta lääkityksen lopetuksesta 49% potilaista oli yhä edelleen ilman lääkehoitoa. Ne tekijät, joilla oli vaikutusta siihen, että hoito voitiin onnistuneesti lopettaa, oli TKE-hoidon pituus ennen lääkkeen lopetusta ja molekulaarisen hoitovasteen syvyys (toisin sanoen kuinka alhainen BCR-ABL1 geenitaso oli PCR-tutkimuksella mitattuna). Ne potilaat, joilla taudin määrä lisääntyi lopetuksen seurauksena, aloittivat TKE-lääkityksen uudelleen ja he kaikki ovat saavuttaneet uudelleen hyvän hoitovasteen.
Tämän uuden tutkimuksen tarkoituksena on selvittää, miten suurella osalla potilaista voidaan hyvä hoitovaste säilyttää toisen TKE-lääkkeenlopetusyrityksen jälkeen. Koska hoitoajan kestolla ja hoitovasteen syvyydellä on merkitystä, oletamme että kahden vuoden hoito tehokkaammalla toisen polven TKE-lääkkeellä (dasatinibi, Sprycel) ennen toista lääkkeenlopetusta voisi lisätä niiden potilaiden määrää, jotka voivat onnistuneesti lopettaa TKE-lääkityksen ja jatkaa ilman lääkehoitoa. Dasatinibi lääke on hyväksytty KML-potilaiden hoitoon sekä ensimmäisessä että toisessa linjassa. Tutkimuksissa on osoitettu, että imatinibiin verrattuna suurempi osa potilaista saa sillä paremman ja syvemmän hoitovasteen.
Tämän lisäksi haluamme tutkia, mitkä tekijät vaikuttavat mahdollisuuteen lopettaa TKE-lääkitys (kuten immuunipuolustusjärjestelmän toiminta) ja lisäksi tutkitaan lääkkeiden käyttöön liittyviä elämänlaadullisia kysymyksiä.
Tutkimukseen otetaan siis tutkittavia, jotka ovat kertaalleen kokeilleet lääketauotusta, mutta ovat joutuneet aloittamaan lääkityksen uudelleen. 1.tauotuskokeilun osalta on tutkittavan pitänyt täyttää EURO-SKI- tutkimuksen sisäänottokriteerit hoidon keston, vasteen ja hoidon muutosten osalta. Tauotuskokeilun päättymisestä on pitänyt kulua 12kk. 1.tauotusta edeltävällä TKE-lääkkeellä ei ole merkitystä eli myös dasatinibia käyttäneet tutkittavat ovat mahdollisia.
EURO-SKI
- Kirjaudu tai rekisteröidy kommentoidaksesi
EURO-SKI
Tutkimuksen tutkittavan tiedote ja suostumus, lupa-asiakirjat, sekä protokolla löytyvät tämän sivun liitteenä (attachment)
Tutkimuksen nimi, lupapäivämäärät, vastuututkija, keskukset
Nimi: Multicenter trial estimating the persistance of molecular remission in chronic myeloid leukaemia after stopping TKI
EudraCT: 2011-000440-22
Eettisen tmk:n/Fimean lupapäivämäärät (tunnisteet):
Toimeksiantaja: Heidelbergin yliopisto, Saksa/European LeukemiaNet
Tutkimuksen vaihe: Faasi II
Tutkimuksen aloituspäivämäärä (FPFV):
Tutkimuksesta vastaava henkilö: Kimmo Porkka
Osallistuvat keskukset (vastuututkija): HUS (Perttu Koskenvesa)
Lyhyt yhteenveto
Etenevä monikeskustutkimus, jossa tutkitaan molekylaarisen remission säilymistä kroonisessa myelooisessa leukemiassa (KML) tyrosiinikinaasin estäjähoidon (TKE) lopetuksen jälkeen
Krooninen myelooinen leukemia (KML) on pahanlaatuinen verisairaus, jonka syntymekanismi tunnetaan hyvin tarkkaan. Sairaudelle tyypillisenä muutoksena todetaan valtaosalla potilaista kromosomien 9 ja 22 välinen translokaatio, jonka seurauksena kromosomissa 9 oleva BCR- geeni ja kromosomin 22 pitkässä haarassa oleva proto-onkogeeni ABL muodostavat sairaudelle tyypillisen fuusiogeenin. Tämä geeni tuottaa BCR-ABL1- onkoproteiinia, jonka tyrosiinikinaasiaktiivisuus aiheuttaa näiden leukeemisten kantasolujen jakautumisaktiviteetin huomattavan kasvun.
Tämän perustavaa laatua olevan havainnon pohjalta onnistuttiin kehittämään kyseisen proteiinin vaikutuksen estäviä lääkkeitä, tyrosiinikinaasin estäjiä (TKE, imatinibi, nilotinibi, dasatinibi), jotka ovat mullistaneet KML:n hoidon. Käytännössä kaikille uusille kroonisen vaiheen potilaille käynnistetään TKE- hoito, joka on lähtökohtaisesti pysyvä. KML todetaan 1-2/100 000 uudella potilaalla vuosittain. Nykyisellään lääkityksen vuosikustannus on 32- 58 000 €/v.
Lääkityksen tehon seurannassa käytetään ensissijaisesti verestä määritettävää herkkää jäännöstautitutkimusta. Siinä BCR-ABL1-fuusiogeenin määrää verrataan elimistössä normaalisti esiintyvän vertailugeenin määrään. Määritys tehdään 3kk välein, kunnes potilaan tulos on toistuvasti enintään 0.1%, (merkittävä molekylaarinen vaste, MMR), minkä jälkeen seurantaväli voi olla 6 kk. Yhden PCR- määrityksen hinta on 350- 500€.
Kyseessä on prospektiivinen, europpalainen monikeskustutkimus, jonka tarkoituksena on selvittää MMR- vasteen säilymistä TKE- hoidon lopettamisen jälkeen. Imatinibihoidettujen osalta on jo olemassa aiempia tutkimustuloksia. Noin 40% ennalta valikoiduista potilaista on voinut lopettaa lääkityksen ilman tautimäärän lisääntymistä. Aiempien tutkimusten perusteella lopettamisen onnistumistodennäköisyyttä lisääviä potilaskohtaisia tekijöitä ovat olleet edeltävän TKE- hoidon kesto, erinomaisen hoitovasteen edeltävä kesto sekä diagnoosivaiheen matala riskiluokka.
Tässä tutkimuksessa on tarkoitus löytää kliinisiä ja biologiasia tekijöitä, jotka vaikuttavat vasteen säilymiseen. Tutkimuksessa seurataan progressiovapaata aikaa ja kokonaiselossaoloaikaa. Potilailta kerätään elämänlaatukyselyillä tietoa lääkityksen lopettamisvaiheessa, lääkkeettömässä vaiheessa sekä mahdollisen lääkkeen uudelleenaloituksen yhteydessä. Merkittävä tutkittava asia on lääkkeen lopettamisen terveys- taloudellinen vaikutus, sillä lääkkeiden korkean hinnan takia on odotettavissa säästöjä huolimatta tiivistettävästä seurannasta. Tällä laajalla tutkimuksella pyritään arvioimaan niiden potilaiden määrää, joiden kohdalla lääkkeenlopetus voisi onnistua. Mahdollisen lääkkeen uudelleenaloituksen jälkeen seurataan aiemman vastetasonsaavuttamiseen kulunutta aikaa.
Tutkimukseen otetaan ≥18- vuotiaitaKML- potilaita, jotka ovat saaneet kroonisen vaiheen hoitoon TKE- lääkitystä monoterapiana tai yhdistelmässä vähintään kolmen vuoden ajan. TKE- lääkityksen vaihto on saanut tapahtua vain sieto- ongelman takia. Edeltävän vuoden ajalta on oltava kolme PCR- tulosta, joissa IS- asteikolla tautimäärä on ≤ 0.01% eikäkertaakaan raja-arvon yli. Tutkimuksen sisäänottovaiheessa jäännöstaudin määrä tutkitaan keskitetysti erikseen määritetyissä laboratorioissa. Suomessa laboratoriona toimii TYKSLAB. Tutkimukseen otettavilta on oltava tiedot diagnoosivaiheen riskiluokitukseen vaadittavista arvoista. Hedelmällisessä iässä olevien naisten on huolehdittava ehkäisystä. Kantasolusiirron läpikäyneitä ei tutkimukseen hyväksytä. Tutkimuksen rekrytointiaika on 2 vuotta ja seurantavaihe kolme vuotta.
Tutkittavia seurataan verenkuvan ja jäännöstaudin osalta kuukausittain ensimmäisen 6 kk ajan, seuraavan 6 kk aikana 6 vkon välein ja sen jälkeen 3 kk välein. Kliininen tutkimus tehdään 3 kk välein. Taudin relapsiksi tulkitaan yksittäinen >0.1% jäännöstautitulos. Siinä tilanteessa potilaalle aloitetaan aiempi TKE- lääke aiemmin käytetyllä annoksella.
Tutkimukseen liittyy useita alatutkimuksia, joilla selvitellään potilaiden immunologisen tilanteen muutoksia lääkemuutoksiin liittyen, pyritään kehittämään herkempää tautimäärän seurantakeinoa ja löytämään vasteen kestoa selittäviä tekijöitä. Näitä varten otetaan säännöllisiä verikokeita. Suomalaiset potilaat osallistuvat käytännössä vain Helsingissä tehtävään pohjoismaiseen immunologiseen tutkimukseen.
Sisäänottoaiheet
- Vähintään 3 vuotta TKE- hoitoa, viimeisin vuosi MR4-tason vasteessa
- Edeltävän 10-14kk ajalta oltava kolme PCR- määritystä, joissa tulostaso enintään 0.01% eikä yhtään sen ylittävää välissä.
- TKE- lääkitystä on voitu vaihtaa vain sivuvaikutusten takia
JATKOSEURANTA: Edelleen lääkkeettömässä seurannassa 36kk kohdalla olevat jatkavat pohjoismaisen ryhmän jatkoseurantaosiossa ad 72kk.
Tiedote/ elokuu 2019
- Kirjaudu tai rekisteröidy kommentoidaksesi
Tiedote/ elokuu 2019
Ohessa lyhyt tietoisku rekrytoivista KML- tutkimuksista elokuussa 2019
Tutkimuksiin osallistuminen ei aiheuta potilaalle tai sairaanhoitopiirille lisäkustannuksia normaaliseurantaan verrattuna. Tutkimuslääkkeet ovat maksuttomia.
Vastatodettu KML-tauti
BosuPeg
- A safety and efficacy study of adding low dose pegylated IFN-αlpha2b to bosutinib in patients with newly diagnosed chronic phase chronic myeloid leukemia
- Pohjoismaisen KML-ryhmän akateeminen tutkimus, mukana myös Ranska
- Tavoitteena Suomesta 15 tutkittavaa, koko tutkimuksessa 212 tutkittavaa.
- Hoito
- 3kk bosutinibimonoterapia (200-300-400 mg/pv), jonka jälkeen arpotumisen perusteella puolelle tutkittavista ro-Peg-IFN-a (Besremi ® (50ug) liitetään lääkitykseen (1krt/ 2 vkoa) s.c.
- Kombinaatioterapian kesto 21 kk (3-24kk), jonka jälkeen monoterapia jatkuu 48kk asti
- Tauotuskokeilun mahdollistavan vasteen saavuttavat tutkittavat lopettavat lääkityksen kohdassa 48kk ja seuranta jatkuu aina 84kk asti.
- Inkluusiokriteerit
- 18-70v, vastadiagnosoitu kroonisen vaiheen KML, ei muita hoitoja vielä (hydroksiurea sallittu)
- Ekskluusio
- Tyypilliset poissulkukriteerit, vaikeat aktiivit autoimmuunitaudit ja depressio
- Muuta
- Tutkimuslääkkeet potilaalla ilmaisia
- Matkakulut korvataan
Hoitoresistentti KML, krooninen ja kiihtynyt vaihe
- ei käynnissä olevia tutkimuksia
Vasteen parantaminen tavoitteena lääkkeen lopetus
DASTOP2
- Etenevä, avoin, yhden hoitohaaran tutkimus, jossa 1. EURO-SKI- kriteerein tehdyssä tauotuskokeilussa epäonnistuneet tutkittavat aloittavat 2 vuoden ajaksi dasatinibilääkityksen ennen uutta tauotuskokeilua.
- 140 tutkittavaa, Pohjoismaat, Saksa, Hollanti
- Hoito
- Dasatinibi 20-100mg 1x1 siedettävyyden mukaan
- MR4-tasoinen vaste saavutettava ja säilyttävä annoslaskuista huolimatta
- 1. vuoden aikana saavutettava MR4-tason vaste, jonka säilyttävä 2.vuoden ajan
- 2v kohdalla vasteen säilyttäneet tutkittavat tauottavat lääkityksen ja seuranta ad 5v
- lääkityksen uudelleenaloitus dasatinibilla, mutta pitkäaikaishoidossa palataan mahdollisuuksien mukaan aiempaan hoitoon
- Inkluusio
- Kroonisen vaiheen KML-potilaat, jotka kertaalleen kokeilleet tauotusta EURO-SKI- kriteerien mukaan, mutta joutuneet aloittamaan lääkityksen uudelleen
- Aiemmalla lääkityksellä jatkettu 1 vuosi tauotuskokeilun päättymisestä
- Ekskluusio
- Lisääntynyt riski dasatinibista aiheutuville haitoille
- Useampi kuin 1 aiempi tauotuskokeilu
- Muuta
- Tutkimuslääkkeet potilaalla ilmaisia
- Matkakulut korvataan
Lääkkeen lopetus
- NCMLSG013 on laboratorionäytteitä tutkiva tutkimus, johon haetaan 1. tauotuskokeilua tekevät EURO-SKI-kriteerit täyttävät tutkittavat
- Tarvitaan SHR- ja HRUHLAB2- suostumukset
- Ei vaikuta muuten tauotuksen toteutukseen, mutta oletuksena on EURO-SKI- tutkimuksen mukainen PCR- seuranta ja lääkityksen aloituskynnys.
Laboratoriotutkimushankkeet
-
Lääkeherkkyyden mittaus
-
Blastikriisivaiheen potilaista teemme in vitro- lääkeherkkyysmittausta mahdollisten uusien lääkehoitojen löytämiseksi
-
Tutkimus on kokeellista tutkimusta ja tällä hetkellä tuloksia ei voi suoraan hyödyntää potilaiden hoitoon
-
-
Näytteet
-
Veri- tai luuydinnäyte potilaasta, jolla todetaan KML:n blastikriisi
-
10-30ml näytettä EDTA putkeen
-
- Muuta
- Näytteen lähetys yms. kulut korvataan tutkimusprojektista
- Ilmoita näytteestä etukäteen hematologiseen tutkimusyksikköön (satu.mustjoki@helsinki.fi)
- Potilaalta tulee pyytää suostumus näytteen analysointia varten (lomake löytyy sivulta: http://www.hematology.fi/hruhlab2) ja SHR- suostumus.
- NCMLSG013
- 1. tauotuskokeilua suunnittelevat
- Veri- ja luuydinnäytteet tauotusta edeltävästi ja mahdollisessa relapsivaiheessa
- Tarvitaan HRUHLAB2 - ja SHR- suostumukset kuten edellä.
- Ei vaikuta muuten potilaan hoitoon, mutta oletus on EURO-SKI- tutkimuksen mukaisen PCR- seurannan toteutuminen.
- Lisätiedot päätutkija Anna Kreutzman (@helsinki.fi) ja tutkimushoitaja Minna Pajuportti (@helsinki.fi)
Helsingissä 30.8.2019
Kollegiaalisin terveisin,
Kirjallisuutta
Kirjallisuutta
- Baccarani et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood.2013;122(6):872-884
| Liite | Koko |
|---|---|
| 599.04 KB |
Potilasohje
- Kirjaudu tai rekisteröidy kommentoidaksesi
Potilasohje
Suomen syöpäpotilaat ry on julkaissut 2012 kolmannen painoksen KML-potilasoppaasta. Oppaan voi ladata PDF muodossa suoraan verkosta tai opasta voi tilata maksutta yhdistyksestä. Lisätietoa KML-potilaille ja heidän omaisilleen on tarjolla Syöpäpotilaat ry:n alaisen KML-potilasverkoston -sivuilla. Perustietoa KML:sta saa myös videomuodossa www.minullatodettiin.fi
Lymfoomat
Lymfoomat
Taudin status
Taudin status
SHR: status-lymphoma
Viite: JCO, Vol 25, No 5 2007: pp. 579-586
|
|
Lyhenne |
Definition |
Nodal Masses |
Spleen, Liver |
|
0 |
Dg |
Diagnoosivaihe |
|
|
|
1 |
CR |
Disappearance of all evidence of disease |
FDG-PET negative; regression to normal size on CT |
Not palpable, nodules disappeared |
|
2 |
PR |
Regression of measuable disease and no new sites |
≥ 50% decrease in SPD of up to 6 largest dominant masses; no increase in size of other nodes |
≥ 50% decrease in SPD of nodules ( |
|
3 |
SD |
Failure to attain CR/PR or PD |
(a) FDG-avid or PET positive prior to therapy; PET positive at prior sites of disease and no new sites on CT or PET |
|
|
4 |
Relapsed disease or PD |
Any new lesion or increase by ≥ 50% of previously involved sites from nadir |
Appearance of a new lesion(s) > 1.5 cm in any axis, ≥ 50% increase in SPD of more than one node, or ≥ 50% increase in longest diameter of a previously identifed node > 1 cm in short axis |
> 50% increase from nadir in the SPD of any previous lesions |
|
9 |
Relapsi (neuro) |
Isoloitu neurorelapsi |
|
|
|
90 |
Exitus-relapsi |
|
|
|
|
91 |
Exitus-infektio |
|
|
|
| 94 | Exitus - ei hoitoa | |||
|
95 |
Exitus, syy tuntematon |
|
|
|
|
999 |
Lost-to-follow-up |
|
|
|
Luokitus
- Kirjaudu tai rekisteröidy kommentoidaksesi
Luokitus
Ann Arbor
Ann Arbor
Lymfoomien levinneisyyden selvittäminen, Ann Arbor / Cotswolds ‑luokitus
Lymfoomien hoitoratkaisut tehdään yleensä levinneisyysasteen selvittämisen jälkeen. Levinneisyys ilmaistaan Ann Arbor/Cotswolds ‑luokituksen, affisioituneiden alueiden luetteloinnin, suurimman tuumorin koon ja pernan pesäkkeiden lukumäärän (Hodgkin-lymfoomissa) perusteella. Hodgkin-lymfoomassa pernan tautipesäkkeiden lukumäärä, yli 4 vs. 4 tai vähemmän on merkittävä ennusteeseen vaikuttava tekijä. Luokittelussa lymfaattista kudosta ovat imusolmukkeet, perna, thymus, Waldeyerin rengas ja Peyerin imukudos suolessa.
Kliininen levinneisyysaste (CS, clinical stage) määritetään anamneesin, statuksen, primaarisen biopsian, luuydinbiopsian ja kuvantamismenetelmien avulla. Patologisen levinneisyysasteen (PS, pathological stage) määritys perustuu lisäksi laparotomian yhteydessä otettuihin näytteisiin.
Aste I:
Lymfooma yhdellä imusolmukealueella tai yhdessä lymfaattisessa elimessä.
Aste II:
Lymfooma kahdella tai useammalla imusolmukealueella samalla puolella palleaa. Affisioituneiden alueiden lukumäärä voidaan osoittaa alaindeksillä, esim II3.
Aste III:
Lymfooma imusolmukkeissa molemmin puolin palleaa. Jos lymfoomaa on pallean yläpuolella, levinneisyysaste III jaetaan kahteen alaryhmään. III1 asteessa lymfoomaa todetaan vain ylävatsalla coeliaca-suonten alueella, pernan portissa, pernassa tai maksan portin alueella. Muualla pallean alapuolella (mm. para-aortaalisissa, parakavaalisissa, iliakaalisissa tai mesenteriaalisissa imusolmukkeissa) oleva tauti luokitellaan III2:ksi.
Aste IV:
Lymfooma disseminoituneena yhdessä tai useammassa ekstralymfaattisessa elimessä tai kudoksessa muuten kuin E-lesiona. Lisäksi lymfooma voi olla imusolmukkeissa. Rajanveto E‑lesion ja stage IV:n välillä on joskus ongelmallista. Suoraa kasvua imusolmukkeesta ympäristön ekstralymfaattiseen kudokseen (esim. lihas, iho, luu) pidetään E‑lesiona. Maksassa tai luuytimessä todettu lymfooma muuttaa levinneisyysluokan aina IV:ksi.
Lisämääreet:
A: Ei yleisoireita
B: Yleisoireet, joiksi luetaan selittämätön yli 38oC kuume, merkittävä yöhikoilu tai yli 10 % painon lasku 6 kuukauden kuluessa. Lyhytaikainen infektioon liittyvä kuume ei ole B‑oire.
X: Suuri tuumori, imusolmukemassan suurin läpimitta yli 10 cm tai mediastinumin leveneminen yli 1/3 thoraxin läpimitasta mitattuna pystyasennossa otetusta kuvasta 5.-6. rintanikaman korkeudelta.
E: Yksittäisen ekstralymfaattisen elimen infiltraatio imusolmukeaffiision vieressä.
Seuranta ja vastearvio
- Kirjaudu tai rekisteröidy kommentoidaksesi
Seuranta ja vastearvio
Potilasohje
- Kirjaudu tai rekisteröidy kommentoidaksesi
Potilasohje
| Liite | Koko |
|---|---|
| 617.63 KB |
MDS
MDS
Myelodysplastiset oireyhtymät:
- klonaalisia hematopoieettisen kantasolun sairauksia, joissa keskeisenä piirteenä on luuytimen tehoton verisolujen muodostus, joka ilmenee verenkuvassa matalina soluarvoina ja luuytimen morfologiassa solujen erilaistumisen ja kypsymisen häiriintymisenä (dysplasioina)
- yleisin pahanlaatuinen verisairaus yli 70-vuotiaiden ikäryhmässä, jossa sen vuosittainen ilmaantuvuus ylittää 20 / 100 000
- diagnoosi perustuu verenkuvaan, luuytimen morfologiaan, histologiaan ja kromosomitutkimukseen; täydentävänä tutkimuksena virtaussytometria, lähitulevaisuudessa todennäköisesti myös molekyyligenetiikka
- matalan riskin taudissa hoidon tavoitteena on korjata anemiasta johtuvia oireita ja parantaa elämänlaatua
- korkean riskin taudissa ennuste on huono (noin yksi vuosi), ja hoidon tavoitteena on estää leukemisoitumista ja pidentää elinikää
- ainoa potentiaalisesti parantava hoito on allogeeninen kantasolujensiirto
- muita hoitoja ovat hematopoieettiset kasvutekijät, immunomoduloivat lääkkeet (lenalidomidi), immunosuppressiiviset lääkkeet (ATG, siklosporiini), hypometyloivat lääkkeet (atsasitidiini), solunsalpaajat ja oireenmukaiset tukihoidot
Käynnissä olevat tutkimukset:
Celgene: A phase 3, multicenter, randomized, double-blind study to compare the efficacy and safety of oral azacitidine plus best supportive care versus placebo plus best supportive care in subjects with red blood cell transfusion-dependent anemia and thrombocytopenia due to IPSS lower-risk myelodysplastic syndromes. Yhteyshenkilö: Freja Ebeling/HUS. Tutkimuksen potilasrekrytointi päättynyt kesällä 2018.
HUS yhteistyössä pohjoismaisen MDS-ryhmän kanssa: Uusien molekyylilääketieteen menetelmien käyttö myelodysplastisen syndrooman diagnostiikassa ja hoitovasteen seurannassa (MDS-MOLE-2015). yhteyshenkilöt: Freja Ebeling, Johanna Illman/HUS
| Kirjoittajat: | Freja Ebeling, Johanna Gustafsson, Annasofia Holopainen, Johanna Illman, Elina Itkonen, Maija Itälä-Remes, Marjut Kauppila, Mikko Keränen, Sirpa Koskela, Outi Laine, Sini Luoma, Eeva Martelin, Mikko Myllymäki, Minna Nyypyy, Tarja-Terttu Pelliniemi, Eira Poikonen, Anu Partanen, Marja Sankelo, Sanna Siitonen, Timo Siitonen, Anri Tienhaara, Sanna Vormisto |
| Viimeisin päivitys: | 17.02.2017 |
| © Suomen Hematologiyhdistys | Suomen Leukemiaryhmä |
Diagnoosi
Diagnoosi
Diagnoosivaiheen tutkimukset
Anamneesi
- aikaisemmat solunsalpaaja- ja sädehoidot, liuotinaltistukset
- käytössä olevat lääkkeet ja luontaistuotteet, tupakointi, alkoholinkäyttö
- vuoto- ja infektiotaipumus
- mahdolliset sukulaisten verisairaudet
- sukulaisten muut merkittävät sairaudet ja poikkeava syöpien esiintyminen (periytyviä luuytimen toimintahäiriöitäkin ajatellen, mm. Fanconin anemia ja telomeropatiat)
- vanhempien (ja isovanhempien) kuoliniät ja -syyt
- hoitovalintoihin vaikuttavat perussairaudet (komorbiditeetti)
- 70-vuotiailta ja nuoremmilta täyssisarusten olemassaolo, ikä ja yleinen terveydentila
Status
- pernan koko
Veri- ja virtsanäytteet
Perusnäytteet
- B-La, P/S-CRP, B-PVK+T+valkosolujen erittelyjakauma, B/E-Retik, P-Haptog, E-Coomb-0, fP-Trfesat (fP-Fe ja fP-Transf), P/S-TfR, P-Ferrit, biologisesti aktiivinen B12-vitamiini, fE-Folaat, P-Krea, P-ALAT, P-AFOS, P-Bil, P-Bil-kj, P-LD, P/S-Alb, S-Prot-Fr, P/S-Uraat, S-B2Miglo, P/S-TSH, S-HIVAgAb, S-HBsAg, S-HBcAb. S-HCVAb, S-ParvAb, U-Perustutkimus (PLV).
Erikoisnäytteet
- S-EPO, jos harkitaan kasvutekijähoitoa (erytropoietiini +- G-CSF)
- leukosyyttien PNH-koe, jos luuydin on hypoplastinen, harkitaan immunosuppressiivista hoitoa tai PNH on erotusdiagnostinen mahdollisuus
- HLADR15, jos harkitaan immunosuppressiivista hoitoa
- plasman lyijypitoisuus harkinnan mukaan
- B-Hb-Fr harkinnan mukaan
- nuorehkoilla potilailla, etenkin familiaarisen sytopenian yhteydessä, harkitaan Fanconin anemian mahdollisuuden selvittämistä (Fanconin anemian geenipaneelitutkimus)
- nuorehkoilla potilailla (ja etenkin, jos keuhkofibroosia tai dyskeratosis congenita -piirteitä): telomeeripituus ja -mutaatiotutkimukset: 1) Uumaja: Kliniskt Genetiskt Laboratorium, 5-10 ml EDTA-verta + lähetelomake, ensin TERT+TERC -mutaatioanalyysi (8000 Skr), jos neg, DKC1- ja TINF2-mutaatioanalyysi (7000 SKr), harkinnan mukaan telomeeripituus (3000 SKr), anna.norberg@vll.se, 2) Bern: Experimental Hematology, 7-15 ml EDTA-verta + lähetelomake, ensin telomeeripituusanalyysi (1708 SFr eli n. 1632 €) ja jos lyhentymää, mutaatioseulonta portaittain (500-5000 SFr), monika.haubitz@insel.ch, (Kansallinen/yhteispohjoismainen BMF-mutaatioseulontapaneeli on pohdittavana, ei vielä rutiinikäytössä, HUS:n HEMAHARV-pkl/Ulla Wartiovaara-Kautto konsultoitavissa.)
- aplastisen anemian kaltaisen taudinkuvan yhteydessä GATA2-vajausta epäiltäessä mutaatioseulonta: Newcastle, Haematological Sciences, Institute of Cellular Medicine, venetia.bigley@ncl.ac.uk (HUS:n HEMAHARV-pkl konsultoitavissa, kts. ed.)
- hyväkuntoisilta alle 70-vuotiailta HLA-1-tyypitysnäyte (Ly-HLA I), jos harkitaan allogeenisen kantasolusiirtohoidon mahdollisuutta
Luuydinnäyte
- ensisijaiset tutkimukset
- pelkkää anemiaa selviteltäessä alkuun vain perifeerisen veren sivelyvalmisteen ja luuydinaspiraatin morfologinen tutkimus rautavärjäyksineen - seulonta
- MDS-epäilystä syytä mainita morfologialähetteessä, jolloin toivotaan myös <5% blastiosuuden tarkempaa määritystä (koska IPSS-R-luokituksessa yhtenä kriteerinä blastiosuus 0-2% tai > 2%)
- jos luuydinmorfologiassa on dysplasiaviitteitä tai jos anemian lisäksi on trombosytopeniaa, neutropeniaa tai perifeerisessä veressä neutrofiilidysplasiaa, tutkitaan aspiraationäytteestä myös karyotyyppi (luuytimen hematologinen kromosomitutkimus) ja jos G-raitatutkimus ei onnistu, suositellaan harkittavaksi kohdennettua interfaasi-FISH-tutkimusta
- luuydinbiopsia (Bm-PAD) solukkuuden, blastiosuuden ja fibroosiasteen tutkimiseksi
- lisätutkimukset
- luuytimen solujen virtaussytometrisessa immunofenotyypityksessä voidaan todeta dysplastinen kypsymisprofiili neutrofiilisarjassa, monosyyteissä tai erytroblasteissa sekä arvioida blastien osuutta ja niiden mahdollisia poikkeavia antigeeni-ilmentymiä sekä laskea ns. Ogata score. Virtaussytometrisesta immunofenotyypityksestä voidaan saada lisätukea MDS:n diagnoosille ja ennusteen arvioimiselle, ja sitä voidaan käyttää apuna hoitovasteen seurannassa. Virtaussytometrinen immunofenotyypitys tehdään harkinnan mukaan, ja eri keskuksissa on erilaiset käytännöt.
- MDS:ssä kuvattujen mutaatioiden seulonta syväsekvensointimenetelmillä (NGS) on tulossa olennaiseksi osaksi MDS-diagnostiikkaa ja ennustearviointia, mutta ei vielä ole kaikkialla rutiinikäytössä eikä toistaiseksi vaadittavissa diagnostiikkaan. Noin 90%:lla MDS-potilaista on kuvattu löytyvän tautimutaatioita, joita on kuvattu >40 myelooisessa geenissä. Yleisimmät mutatoituneet geenit ovat TET2, SF3B1, ASXL1, DNMT3, SRSF2, RUNX1, U2AF1, EZH2 ja TP53. Mutaatiotiedon soveltaminen kliinisiin ratkaisuihin on nopeassa kehitysvaiheessa.
Erotusdiagnostiikassa huomioitavaa
- B12-vitamiinin tai folaatin puute
- kroonisen sairauden anemia
- autoimmuunisytopeniat
- krooninen maksasairaus ja/tai runsas alkoholinkäyttö
- krooniset virusinfektiot (HIV/HBV/HCV/Parvo/CMV/EBV)
- raskasmetallialtistukset
- lääkeaineiden tai muiden toksisten aineiden aiheuttamat sytopeniat
- aplastinen anemia
- PNH
- myelofibroosi
- muut kantasolusairaudet (mm. akuutit leukemiat)
- Fanconin anemia
- LGL-leukemia, karvasoluleukemia
- geneettiset ei-malignit tilat, kuten kongenitaalinen sideroblastianemia
Luokitus
Luokitus
- FAB-luokitus, jossa myelodysplastinen oireyhtymä (MDS) jaettiin viiteen alaryhmään blastiosuuden, luuytimen rengassideroblastien ja veren monosyyttimäärän perusteella, oli käytössä vuodesta 1982.
- WHO 2008 -luokitus, joka on esitetty alla olevassa taulukossa.
- WHO 2008 -luokitukseen 2016 tehdyt revisiot, joista lisätty lyhyet maininnat kursiivilla punaisella fontilla allaolevaan taulukkoon.
- FAB-luokitukseen perustuva MDS-riskiluokitus, IPSS-ennusteluokitus (International Prognostic Scoring System) julkaistiin 1997 (taulukossa, joka löytyy seuraavalta sivulta, myös keskimääräisiä elinaikatietoja).
- Uusi, 2012 julkaistu IPSS-R-luokitus jakaa MDS-potilaat viiteen ennusteeltaan eroavaan ryhmään ja on tulossa yleiseen käyttöön. Siinä erotellaan sytopenioiden syvyys aiempaa tarkemmin samoin kuin luuytimen blastiosuus (0-2% ja >2-5%), ja sytogenetiikan perusteella erotellaan aiemman kolmen asemasta viisi riskiryhmää. (IPSS-R-riskipistelaskuri: http://ipss-r.com/)
-
WHO-luokitukseen pohjautuva WPSS-ennusteluokitus julkaistiin 2007 ja soveltuu käytettäväksi myös diagnoosivaiheen jälkeen.
- Uusia, vielä arvioitavana olevia mahdollisia ennustetekijöitä ovat ferritiinitaso, transfuusiotiheys, luuytimen fibroosi, LD-taso, beeta-2-mikroglobuliinitaso ja CD34-soluklusterit luuytimessä.
- ei-hematologisten oheissairauksien huomioimiseen riskiarviossa on kehitetty MDS-specific comorbidity index (MDS-CI) (Della Porta et al. 2011)
Taulukko. WHO 2008 -luokitus, johon lisätty myös ICD-0-koodit, punaisella 2016 revisiot
|
Alaluokka |
Myeloblastit veressä (%) |
Muu |
Myeloblastit ytimessä (%) |
Luuytimen löydökset |
|
RCUD 2016: MDS-SLD ilman erittelyä |
<11 |
yhden, joskus kahden linjan sytopenia |
< 5 |
>10 % yhden linjan soluista dysplastisia <15% erytroblasteista rengassideroblasteja |
|
RARS 2016: MDS-RS-SLD
|
0 |
anemia |
< 5 |
>15% erytroblasteista rengassideroblasteja, 2016: >=5 % jos SF3B1 -mutaatio vain erytroisen linjan dysplasia
|
|
RCMD 2016: MDS-MLD tai MDS-RS-MLD |
<11 |
sytopeniat |
< 5 |
>10 % kahden tai useamman linjan soluista dysplastisia, ei Auerin sauvoja ±15% rengassideroblasteja 2016: >=5% jos SF3B1 -mutaatio |
|
RAEB-1 2016: MDS-EB-1 |
<51,2 |
sytopeniat |
5-92 |
yhdessä tai useammassa linjassa dysplasiaa, ei Auerin sauvoja 2016: 0-3 dysplastista linjaa |
|
RAEB-2 2016: MDS-EB-2 |
5-19 |
sytopeniat |
10–19 |
yhdessä tai useammassa linjassa dysplasiaa, Auerin sauvoja voi olla 2016: 0-3 dysplastista linjaa |
|
MDS-U |
<11 |
sytopeniat |
< 5 |
MDS:aan viittaavaan kromosomipoikkeamaan (kts. Table 6 linkkinä olevassa kirjallisuusviitteessä) liittyvänä hyväksytään <10 %:n dysplasia yhdessä tai useammassa linjassa |
|
MDS |
<1 |
anemia, |
< 5 |
megakaryosyyteissä aliliuskoittumista, ei Auerin sauvoja 2016: del(5q) alone or with 1 additional abnormality except -7 or del(7q) |
WHO 2016: Arber DA et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016:127:2391-2405. (https://www.ncbi.nlm.nih.gov/pubmed/27069254)
Brunning RD, Porwit A, Orazi A, ym. Myelodysplastic syndromes. Kirjassa: Swerdlow SH, Campo E, Harris NL, toim. WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. Lyon: IARC, 2008; 88-107.
1. Jos veressä on blasteja 2-4 %, luokitus on RAEB-1, vaikka luuytimen blastiosuus on < 5 %.
Jos veressä on blasteja 1 %, luokitus on MDS-U.
2. Auerin sauvojen esiintyminen vie luokituksen RAEB-2:een
RCUD refraktaarinen sytopenia, jossa yhden linjan dysplasia
RA refraktaarinen anemia
RN refraktaarinen neutropenia
RT refraktaarinen trombosytopenia
RARS refraktaarinen anemia, jossa rengassideroblasteja
RCMD refraktaarinen anemia, jossa monilinjainen dysplasia
RAEB-1 refraktaarinen anemia, jossa blastiylimäärä-1
RAEB-2 refraktaarinen anemia, jossa blastiylimäärä-2
MDS-U luokittelematon MDS
MDS del(5q) MDS, jossa ainoana muutoksena 5q-
MDS-SLD MDS with single lineage dysplasia
MDS-MLD MDS with multilineage dysplasia
MDS-RS MDS with ring sideroblasts
MDS-RS-SLD MDS-RS with single lineage dysplasia
MDS-RS-MLD MDS-RS with multilineage dysplasia
MDS-EB MDS with excess blasts
MDS-U MDS, unclassifiable
IPSS
IPSS
| 0 | 0,5 | 1 | 1,5 | 2 | |
| Luuytimen blastimäärä (%) | <5 | 5-10 | - | 11-20 | 21-30** |
| Karyotyyppi |
Pieni riski normaali -Y ainoana 5q- ainoana 20q- ainoana
|
Keskikorkea riski muut poikkeavuudet |
Suuri riski ≥ 3 poikkeavuutta kromosomin 7 mikä tahansa poikkeavuus
|
||
| Sytopeniat* | ei yhtään tai 1 | 2 tai 3 | |||
|
*Hb<100 g/L; B-Neutr<1,5x109/L; tromb<100x109/L **Nykyisen WHO-luokituksen mukaan ei enää MDS, vaan akuutti leukemia |
|||||
Keskimääräinen elinaika diagnoosista ja aika transformaatioon AML:ksi (vuosia)
| IPSS-pisteet | Riskiluokka | Potilaita (%) |
Ikä ≤ 70v Elinaika |
Ikä > 70v Elinaika |
Aika, jossa 25%:lle AML-evoluutio (v) |
| 0 | matala | 31 | 9,0 | 3,9 | 9,4 |
| 0,5-1 | keskikorkea-1 | 39 | 4,4 | 2,4 | 3,3 |
| 1,5-2 | keskikorkea-2 | 22 | 1,3 | 1,2 | 1,1 |
| ≥ 2,5 | korkea | 8 | 0,4 | 0,4 | 0,2 |
WPSS-ennusteluokitus
|
|||||||||||||||||||||||||
NOTE. Risk groups were as follows: very low (score = 0), low (score = 1), intermediate (score = 2), high (score = 3 to 4), and very high (score = 5 to 6).
Abbreviations: MDS, myelodysplastic syndrome; RA, refractory anemia; RARS, refractory anemia with ringed sideroblasts; 5q–, myelodysplastic syndrome with isolated del(5q) and marrow blasts less than 5%; RCMD, refractory cytopenia with multilineage dysplasia; RCMD-RS, refractory cytopenia with multilineage dysplasia and ringed sideroblasts; RAEB-1, refractory anemia with excess of blasts-1; RAEB-2, refractory anemia with excess of blasts-2.
* Karyotype was as follows: good: normal, –Y, del(5q), del(20q); poor: complex (≥ three abnormalities), chromosome 7 anomalies; and intermediate: other abnormalities.
† RBC transfusion dependency was defined as having at least one RBC transfusion every 8 weeks over a period of 4 months.
Seuranta ja vastearvio
Seuranta ja vastearvio
Seuranta
Matalamman riskiryhmän MDS (IPSS matala ja keskikorkea-1)
- Oireetonta potilasta seurataan verenkuvan avulla 2-4 kk välein. Etenkin iäkkäiden potilaiden seuranta voidaan toteuttaa terveyskeskuksessa tai aluesairaalassa. Potilaita, joiden kohdalla allogeeninen kantasolujensiirto voi tulla kyseeseen, seurataan keskussairaalassa tai yliopistollisessa keskussairaalassa.
Korkeamman riskiryhmän MDS (IPSS keskikorkea-2 ja korkea)
- Korkeamman riskin potilaille tulee harkita hoidon aloitusta, ja seuranta on siten yksilöllistä (kts. Hoito-osio jäljempänä).
Hoidon aloittamisaiheet:
- oireinen sytopenia (yleensä anemia, Hb < 100 g/l)
- suureneva ytimen blastiosuus (>10-15%)
- korkeamman riskin tauti (IPSS keskikorkea-2 ja korkea)
Hoitovasteen kriteerit MDS:ssa International Working Groupin (IWG) mukaan
(CR- tai PR-vasteen keston oltava korkeamman riskin MDS:ssa vähintään 4 viikkoa, joskaan 1 kk kuluttua otettavaa luuydinnäytettä ei välttämättä vaadita. Matalamman riskin MDS:ssä vasteen (lähinnä HI) tulisi kestää vähintään 8 viikkoa.)
|
Täydellinen vaste (CR)
|
Myeloblasteja < 5 % luuytimen soluista, kypsyminen kaikilla solulinjoilla normaalia. Solumorfologia voi olla dysplastista. Verenkuva:
|
|
Osittainen vaste (PR) |
CR:n kriteerit täyttyvät kaikkien ennen hoitoa todettujen poikkeavien löydösten osalta paitsi
Solukkuudella tai morfologialla ei ole merkitystä. |
|
Sytogeneettinen vaste |
Täydellinen vaste
Osittainen vaste
|
|
Verenkuva-arvojen korjaantuminen (HI) (lähtötaso kahden hoitoa edeltävän, vähintään viikon välein mitatun, lukeman keskiarvo) (matalamman riskin MDS:ssä korjaantuminen vähintään 8 viikoksi) |
Hemoglobiinivaste, lähtötaso < 110 g/l
Trombosyyttivaste
Neutrofiilivaste, lähtötaso < 1.0x109/l
|
|
Stabiili tauti |
PR-vastetta ei ole saavutettu, mutta tauti ei ole edennyt >8 viikon aikana |
|
Epäonnistunut hoito (failure) |
Hoidon aikainen kuolema tai taudin eteneminen, mikä ilmenee sytopenioiden syvenemisenä, luuytimen blastiosuuden lisääntymisenä tai taudin etenemisenä korkeamman riskin FAB-alaryhmään. |
|
Taudin eteneminen |
Muutos luuytimen blastimäärässä, kun lähtötaso
Jokin seuraavista:
|
|
Relapsi täydellisestä tai osittaisesta vasteesta |
Vähintään yksi seuraavista
|
Cheson BD, Greenberg PL, Bennett JM, ym. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419-25.
Hoitovasteen arviointi
Diagnoosivaiheessa tehtyjen tutkimusten perusteella valitaan potilaskohtaiset seurantatutkimukset (sytogenetiikka, molekyyligenetiikka, immunofenotyypitys), mikäli sellaisia on olemassa.
Taudin status
Taudin status
Vastearvion yhteydessä pyritään tautitaakan arvio ja taudin tilanne luokittelemaan oheisen taulukon mukaisesti (SHR-muuttuja: status-mds). Taulukosta valitaan arviointihetkellä oleva paras vaste.
|
Koodi |
Lyhenne |
Nimike |
Määritelmä |
|
0 |
Dg |
Diagnoosivaihe |
|
|
1 |
CR-MRDneg |
Remissio, jäännostauti negatiivinen |
|
|
2 |
CR-MRDpos |
Remissio, jäännostauti positiivinen |
|
|
3 |
CCR |
Sytogeneettinen remissio (complete cytogenetic response) |
|
|
4 |
pCR |
Osittainen sytogeneettinen vaste (partial cytogenetic response) |
|
|
5 |
CR |
Morfologinen remissio (complete remission) |
|
|
6 |
PR |
Osittainen vaste (partial remission) |
(esimerkki: blastiosuus 15%-> 6%)
|
|
7 |
HI |
Verenkuva-arvojen korjaantuminen (hematologic improvement) (lähtötaso kahden hoitoa edeltävän, vähintään viikon välein mitatun, lukeman keskiarvo) (matalamman riskin MDS:ssä korjaantuminen vähntään 8 viikoksi) |
(esim: B-neut 0,3-> 0,6) |
|
8 |
Stabiili |
Stabiili tauti (stable disease) |
|
|
9 |
Progressio |
Taudin eteneminen (progressive disease) |
|
|
10 |
Relapsi |
Relapsi täydellisestä tai osittaisesta vasteesta |
|
|
90 |
Exitus-progressio |
Exitus, progressio |
|
|
91 |
Exitus-infektio |
Exitus, infektio |
|
|
92 |
Exitus-muu |
Exitus, muu syy |
|
|
99 |
Exitus-tuntematon |
Exitus, syy tuntematon |
|
|
999 |
Poistunut seurannasta |
Lost to follow-up |
|
Hoito
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hoito
- Hoidon valintaan vaikuttavat IPSS-luokitus (matalampi tai korkeampi riski), potilaan ikä ja hänen liitännäissairautensa ja yleinen raihnaisuuden (fragiliteetin) aste.
- Mikään lääkehoidoista ei ole kuratiivinen, jonka vuoksi potilaan kanssa tulee puhua hoidon tavoitteista
- Matalamman riskin taudeissa hoidon tavoitteina ovat sytopenioista johtuvien oireiden korjaaminen ja elämänlaadun parantaminen. Ensilinjan hoitona anemiaan käytetään yleensä kasvutekijähoitoa (erytropoietiini +- G-CSF) ja pienellä osalla potilaista immunosuppressiivista hoitoa. Toisen linjan hoitona tulee 5q- -oireyhtymässä harkittavaksi lenalidomidi ja muilla joskus atsasitidiinihoito, joskaan sillä ei ole virallista käyttöaihetta matalamman riskin MDS:ssä. Osalla hoitoresistenteistä potilaista tai etenevässä taudinkuvassa harkitaan allogeenista kantasolujensiirtoa. Tukihoitoja käytetään muiden hoitojen rinnalla, joskus etenkin iäkkäillä yksinomaisena hoitona.
- Korkeamman riskin taudeissa selvitetään heti dg-vaiheessa allogeenisen kantasolujensiirron mahdollisuus huomioiden potilaan ikä ja liitännäissairaudet. Jos siirto ei tule kyseeseen, ensisijainen hoito on atsasitidiini, jota käytetään tarvittaessa myös kantasolujensiirtoa edeltävästi. Joskus käytetään intensiivistä solunsalpaajahoitoa, mutta matala-annoksista solunsalpaajahoitoa nykyään harvemmin. Hoitojen tavoitteena on elossaoloennusteen parantaminen ja leukemisoitumisriskin pienentäminen.
- Ennusteellisesti merkittävien mutaatioiden osoittaminen saattaa tulevaisuudessa osaltaan ohjata hoitovalintaa.
- vastearviossa auttavat International Working Groupin (IWG) kriteerit (tähän sivuston sisäinen linkki suomenkieliseen taulukkoon, tämä kirjallisuuslinkki siirretään taulukon otsikkoon)
1. Matalamman riskin (IPSS Score ≤ 1.0 tai IPSS-R Score ≤ 4.5) hoidot:

1.1 Sytopenioiden tukihoidot
1.1.1 Anemia
- anemiaan liittyy huonontunut elämänlaatu ja suurentunut sydäntapahtumavaara
- punasolujensiirrot oireiseen anemiaan (B-Hb<80-90 g/l) huomioiden se, että ne aiheuttavat rautakuorman kertymistä
- seuranta: B-PVK+T tarpeen mukaan, P-Ferrit ja transfuusiotarpeen rekisteröinti 3 kk välein
1.1.2 Trombosytopenia
- trombosyyttiensiirtoja vain merkittäviin vuotoihin, ei ennaltaehkäisevästi (poikkeuksena toimenpiteet ja MDS:n hoitoihin liittyvät trombosytopeniat)
- traneksaamihaposta voi olla hyötyä vuotojen ehkäisyssä ja hoidossa
- trombopoietiiniagonisteja tutkittu vaihtelevin tuloksin, Cochrane-katsauksessa (Desborough M et al. Cochrane Database Syst Rev 2016:2016) ei riittävää osoitusta romiplostiimin tai eltrombopagin suosittelemiselle
- eltrombopagia (suuremmilla annoksilla kuin ITP:ssä) voidaan harkita matalamman riskin MDS:ssa ongelmallisen trombosytopenian hoidossa, kun Bm-blastit <5%, mutta ei ole virallinen käyttöaihe
1.1.3 Infektiot
- infektioiden nopea ja tehokas hoito on keskeistä
- rutiininomaisesta valkosolukasvutekijäprofylaksista ei hyötyä oireettomalla neutropeniapotilaalla
- käytetään hoidettaessa neutropeenisen potilaan infektioita, toistuvien vakavien infektioiden estohoidossa ja syvän neutropenian estohoitona atsasitidiinia saavilla (joskin neutrofiilitasolla >0,5 infektioita esiintyy harvoin)
- potilaat tulisi rokottaa pneumokokkia ja influenssaa vastaan ja ohjeistaa hyvään käsihygieniaan
1.2 Muut tukihoidot
1.2.1 Raudan kelaatiohoito
Indikaatio:
- jatkuva punasolujensiirtoja vaativa anemia ja
- odotettavissa oleva elinaika >2 vuotta (eli IPSS matalamman riskin tauti) sekä
- P-Ferrit > 1500 µg/l (yleensä noin 25 punasoluyksikön annon jälkeen)
- Maksan MRI T2* -kuvaus valikoiduissa tapauksissa maksan rautavarastojen määrittämiseksi
- allosiirtoon menevillä varhaisessa vaiheessa, jo estämään ennalta rautakuorman kehittymistä
Toteutus:
Deferasiroksi (ensisijainen)
- aloitusannos 5-10 mg/kg kerran päivässä, nosto asteittain ad 20-40 mg/kg/vrk (tyhjään mahaan vähintään 30 min ennen ateriaa)
- ruoansulatuskanavan haittavaikutuksia ja kreatiniinin suurenemista 25%:lle
- kreatiniinin seuranta alkuun viikottain kuukauden ajan, myöhemmin kuukausittain
- jos kreatiniini suurenee ad 2x viitevälin yläraja, tauotus ja jatkossa aloitus pienemmällä annoksella
- lääkkeen ottaminen ruoan kanssa vähentää vatsaoireita ja ripulia
- Novartiksen edustajan mukaan dispergoituva tabletti korvautuu keväästä 2017 lähtien kalvopäällysteisellä nieltävällä kapselilla, jonka GI-siedettävyys on parempi
Desferrioksamiini
- 5-7 tunnin jatkuva infuusio pumpulla ihon alle, 5-7 vrk/viikko
- annos yleensä 20-60 mg/kg/vrk
- C-vitamiinilisä 1 kk hoidon aloituksesta (max 200 mg/vrk), jollei sydämen vajaatoimintaa
Tavoite ja hoitovaste: P-Ferrit-tavoite kelaatiohoidossa on < 1000 µg/l. Korkeaan ferritiiniin on todettu liittyvän lyhentynyt elinaika. Ei kuitenkaan tiedetä, pidentääkö raudan kelaatiohoidolla aikaansaatu plasman ferritiinitason pieneneminen elinaikaa.
1.2.2 Kasvutekijähoito erytropoietiinivalmisteilla
Indikaatio:
- oireinen anemia (Hb < 100 g/l) matalamman riskin raudissa (Bm-blastit <10%), kun muut anemian syyt poissuljettu ja S-EPO < 500 U/l ja/tai punasolujensiirtotarve < 2 yks/kk
Toteutus:
- epoetiini: aloitusannos on 30 000 KY/vk sc (450 KY/kg/vk) . Jos Hb ei suurene vähintään 15 g/l kahdeksassa viikossa, annos kaksinkertaistetaan
- darbepoetiini: yleensä 500 ug sc kolmen viikon välein tai 300 ug kahden viikon välein. Jos Hb ei suurene vähintään 15 g/l kahdeksassa viikossa, tihennetään annostusta ad 300 ug kerran viikossa
- pienipainoisilla tai munuaisten vajaatoiminnassa annokset yleensä pienemmät
- ellei kaksinkertaistetullekaan EPO-annokselle vastetta, hoitoon voidaan harkita liitettäväksi matala-annoksinen G-CSF, esimerkiksi filgrastiimi 12-30 MU (120-300 ug) 2-3 kertaa viikossa sc. Valkosolukasvutekijähoidossa pyritään selkeään neutrofiilinousuun, tasolle 6-10x109/l
- RARS:ssa aloitetaan suoraan EPO + G-CSF -yhdistelmällä (?, tarkistetaan tulossa olevasta pohjoismaisen ohjeen päivityksestä, ohjeistetaanko vielä näin, ELN ei ohjeista)
- seuraa harvakseltaan ferritiiniarvoa: lisää rautalääkitys, jos ferritiini matala tai fP-Trfesat < 20%
- jos Hb suurenee > 120 g/l, pienennetään annoksia tai harvennetaan pistoksia (tarvittaessa tauotus ja uudelleenaloitus 50%:n annoksella, kun Hb <120 g/l)
Hoitovaste:
- korjaa anemiaa noin puolella potilaista, parantaa elämänlaatua, saattaa pidentää elinaikaa, myöhentää transfuusiotarpeen alkamista
- vaste saavutetaan yleensä 12 viikossa, mutta viimeistään 16 viikossa, vasteen keston mediaani noin 2 vuotta
- jos vastetta ei saavuteta (joko punasolusiirtoriippumattomuus tai stabiilissa anemiassa Hb:n suureneminen >15 g/l), hoito lopetetaan tehottomana ja harkitaan luuydinmorfologian tutkimista mahdolisen progression varalta
1.3 Immunosuppressiiviset lääkkeet
Osalla RA- tai RCMD-potilaista sytopenioiden taustan arvellaan olevan immunologinen, samantyyppinen kuin aplastisessa anemiassa.
Indikaatio
- erytropoietiinihoidolle resistentit matalan riskin alle 60 (-70) -vuotiaat potilaat, joilla on vaikea sytopenia, hypoplastinen ydin, ei blastiylimäärää eikä korkean riskin kromosomipoikkeavuuksia
- HLA DR15 -positiivisuus tai PNH-klooni lisäävät vasteen todennäköisyyttä ainakin yli 50-vuotiailla
Toteutus
- antilymfosyyttiglobuliini (ATG) yksin tai yhdessä siklosporiinin kanssa
- aplastisessa anemiassa hevosen ATG (Atgam®) 40 mg/kg/vrk 4 vrk:n ajan tutkitusti tehokkain, mutta MDS:ssa mahdollista antaa vaihtoehtoisesti kanin ATG (Thymoglobuline®) 3,75 mg/kg/pv päivinä 1-5 tai hevosen ATG (Lymphoglobulin®) 15 mg/kg/pv päivinä 1-4
- kombinoidaan siklosporiini 3-5 mg/kg/vrk jaettuna kahteen osaan (B-CyA -tavoite 150-250 µg/l) vähintään 3-6 kk ajan, hoitoon vastaavassa taudissa usein pitkäaikaisena ylläpitohoitona
- seerumitaudin ehkäisemiseksi prednisoloni 1 mg/kg/vrk päivinä 1-10 asteittain päivinä 11-24 purkaen
- iäkkäillä voidaan harkita siklosporiinia monoterapiana ATG:n toksisuuden vuoksi
- pneumokystisprofylaksi 6 kk
- vaihtoehtona ATG:lle voidaan joskus harkita alemtutsumabia 10 mg/pv iv 10 pv (Neukirchen J et al. Ann Hematol 2014;93:65-9)
Hoitovaste antilymfosyyttiglobuliinihoidolle
- vaste 30-50%:lle
- anemian lisäksi myös muissa sytopenioissa korjautumista
- ilmenee keskimäärin 2,5 kuukaudessa, voi tulla vasta 3-6 kk:ssa
- vasteen kesto keskimäärin 3 vuotta
1.4 Lenalidomidi
Indikaatiot
- FDA on hyväksynyt lenalidomidille käyttöindikaation matalamman riskin MDS:ssä, jossa on kromosomipoikkeavuutena 5q- joko yksin tai muiden poikkeavuuksien kanssa
- EMEA on hyväksynyt lenalidomidin punasolusiirtoriippuvaisille pienen tai Int-1-riskiryhmän MDS-potilaille, joilla 5q- on yksittäisenä sytogeneettisena poikkeavuutena, kun muut hoitovaihtoehdot ovat riittämättömiä tai eivät sovi potilaan hoitoon
- jos luuydinbiopsian immunohistokemiassa voimakas tumien p53-värjäytyminen, on AML-progressioriski suurempi, MDS-004-tutkimuksessa 27.5%/2 vuotta (versus 3.6% IHC-p53-negatiivisilla)
- ei korvattavuutta Suomessa MDS-indikaatiolla (vuonna 2016)
Toteutus
- ennen hoitoa tulisi tutkia TP53-geenin mutaatiostatus joko luuydinbiopsian immunohistokemialla tai sekvensoinnilla; mutaatio todetaan 20%:lla MDS 5q- -potilaista, näillä potilailla taudin ennuste huonompi ja lenalidomidivasteet lyhyempiä
- aloitusannos 10 mg/pv päivinä 1-21 (10 mg/pv annos tehokkaampi kuin 5 mg), syklien pituus 28 pv
- annosta pienennettävä munuaisten vajaatoiminnassa
- alkuvaiheen neutro- ja trombosytopenioita n. 50%:lla, esiintyminen ennustaa hyvää hoitovastetta
- kahden ensimmäisen hoitosyklin aikana verenkuvaseuranta viikottain
- tarvittaessa annosreduktioharkinta, G-CSF-harkinta
- rutiininomaista tromboosiprofylaksiaa ei tarvita
Hoitovaste
- 5q- oireyhtymässä 67% punasolujensiirroista riippumattomiksi, 45%:lle täydellinen sytogeneettinen vaste
- vasteen ilmaantumisajan mediaani 4.4 viikkoa, vastearvioon tarvitaan vähintään 3-4 sykliä
- hoitoon vastaavilla hoitoa jatketaan, vasteen kesto keskimäärin 2,2 vuotta
- täydellisessä sytogeneettisessä vasteessa lopettaneilla hoitovaste on voinut säilyä ilman lääkettäkin
- jatkoseuranta: B-PVK+T+Neutrofiilit 3 kk välein, luuytimen hematologinen kromosomitutkimus 6 kk välein
- satunnaistetussa non-5q- MDS-tutkimuksessa 26%:lle potilaista punasolusiirtoriippumattomuus, vasteen mediaanikesto 41 viikkoa (Raza et al. Blood 2008;111:86-93)
1.5 Allogeeninen kantasolujensiirto
Indikaatiot
- < 70-vuotias hyväkuntoinen matalamman riskin potilas, jolla muuhun hoitoon reagoimaton vaikea sytopenia, huonon ennusteen kromosomilöydös tai suureneva ytimen blastiosuus
- IPSS-R-luokitus voi antaa lisätietoa riskistä; sen keskiriskiryhmässä mutaatioanalyysi saattaa lähitulevaisuudessa auttaa siirtoon menevien potilaiden valinnassa (TP53, ASXL1, RUNX1 ja EZH2 liittyvät huonoon ennusteeseen MDS:ssa
- toteutus ja hoidon vaste: kts. korkeamman riskin hoidot: allogeeninen kantasolujensiirto
- konsultoidaan kantasolujensiirtokeskusta
1.6 Atsasitidiini
- kts. indikaatiot korkeamman riskin kohdalta
- voidaan harkita, jos EPO-hoito ja/tai immunosuppressiivinen hoito eivät tehoa tai tule kyseeseen eikä potilas sovellu allogeeniseen kantasolujensiirtoon (kts. siirtoindikaatiot kohdasta Allogeeninen kantasolujensiirto) etenkin jos potilaalla on vaikea sytopenia tai korkean riskin kromosomilöydös tai blastiylimäärä lisääntyy
- annos vakioannos 75 mg/m2 sc seitsemän pv ajan 28 pv:n sykleissä, myös 5 vrk:n annostelua voi harkita (Lyons RM et al. JCO 2009)
- EPO-resistenteillä 40-50%:lle vaste anemiaan; elinaikahyötyä ei ole toistaiseksi osoitettu
2. Korkeamman riskin (IPSS >/= 1,5 tai IPSS-R >/=4,5) hoidot:
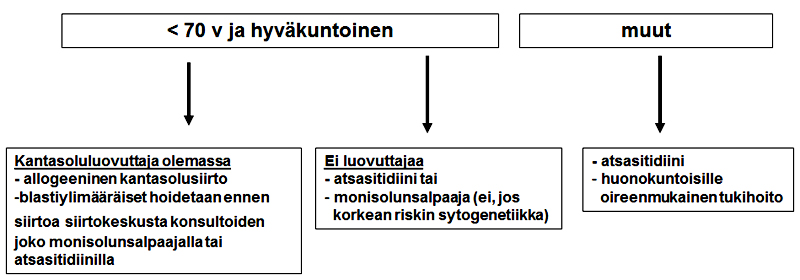
2.1 Hypometyloivat hoidot
2.1.1 Atsasitidiini
- hypometyloiva lääke, joka indusoi kasvuun, erilaistumiseen ja apoptoosiin liittyviä geenejä
- ei paranna MDS:aa, mutta hidastaa sen kulkua
- korkeamman riskin taudissa ensilinjan hoito nillä, joilla ei suoraan edetä kantasolujensiirtoon
- ainoa lääke, jolla on faasin III tutkimuksessa osoitettu elinajan piteneminen MDS:ssa verrattuna tukihoitoon, matala-annoksiseen solunsalpaajahoitoon tai intensiiviseen solunsalpaajahoitoon
Indikaatio
- FDA hyväksynyt käyttöaiheeksi matalamman ja korkeamman riskin MDS:n, mutta EMEA ainoastaan korkeamman riskin MDS:n (ja AML:n)
- voidaan käyttää myös ennen allogeenista kantasolujensiirtoa pienentämään ytimen korkeaa blastiosuutta, etenkin potilailla, joilla on korkean riskin kromosomipoikkeavuus (ei ole virallinen indikaatio)
- 75 mg/m2/vrk sc 7 pv kuurein, syklin pituus 28 vrk
- epävirallisesti käytetään myös annostelua 75mg/m2/vrk päiväaikataululla 5-0-2 (=viikonlopputauko) tai 100 mg/m2/vrk 5 vuorokauden ajan, mutta näiden annostelutapojen tehosta ei ole vertailua vakioannostukseen
- vaste tulee hitaasti, suositellaan (4)-6 hoitojaksoa ennen vastearviota luuytimestä
- vasteista 50% saavutetaan kolmen, 80% kuuden ja 90% yhdeksän syklin jälkeen
- jos saavutetaan vaste, hoitoa suositellaan jatkettavaksi tautiprogressioon saakka
- haittavaikutuksia eniten kahden ensimmäisen syklin aikana
- myelosuppressio, pahoinvointi, GI-oireet, injektiokohdan ärsytys (lievitettävissä helokkiöljyllä tai tulehduskipulääkegeelillä)
- verenkuvakontrolli viikoittain 3 ensimmäisen syklin aikana, sen jälkeen 2 vk välein
- alkuvaiheen sytopenioista huolimatta ei annosreduktioita eikä mielellään myöskään hoitovälien pidennyksiä kolmen ensimmäisen syklin aikana, etenkään edenneessä MDS:ssa
- myöhempien sytopenioiden yhteydessä luuydintutkimus: jos myelosuppressiota ilman viitteitä tautiprogressiosta, annosreduktio esim. ad 50 mg/m2/vrk tai hoitoväli 5 viikkoon
Vaste
- 50%:lla hoitovaste, jonka kesto keskimäärin 13,6 kk
- punasolujen siirtotarve lakkaa 45%:lla
- trombosyyttiluvut korjautuvat 20-30%:lla
- CR- ja PR -vasteita 11-29%:lla
- pidensi elinaikaa faasin III tutkimuksessa 9,5 kk
- kaikentyyppiset hoitovasteet assosioituivat elinaikaetuun
- taudin leukemisoituminen hidastui
- vaste ei riippunut potilaan iästä (myös >75-vuotiaat hyötyivät), FAB/WHO -tyypistä, ytimen blastimäärästä tai karyotyypistä (myös -7/del7q -potilaat vastasivat hoidolle)
- haittavaikutukset kohtuullisen vähäisiä ja infektioita < 20%:lla
- vaste menetetään puolella potilaista 1-1,5 v kuluessa
- kun tauti etenee/relapsoituu, ennuste huono (keskimäärin vain 6.5 kk)
- progressioon/relapsiin ei tällä hetkellä hyviä hoitovaihtoehtoja
2.1.2 Desitabiini
- vasteet samanlaisia kuin atsasitidiinilla korkeamman riskin taudissa, mutta elinaikaetua ei ole osoitettu
- FDA hyväksynyt MDS-indikaatiolla; EMEA ainoastaan AML-indikaatiolla
- annos 20 mg/m2 5 pv (20 mg/m2 10 pv:n annostuksella saatu hyviä vasteita korkean riskin kromosomi- ja TP53-muutoksissa)
- vaihto atsasitidiinista desitabiiniin harvoin hyödyllistä
2.2 Intensiivinen solunsalpaajahoito
- yleisimmin antrasykliini-AraC -yhdistelmä (sytarabiini joko jatkuvana tai kahdesti päivässä)
Indikaatio
- <65-vuotiaille korkeamman riskin potilaille
- vastetta ennustavat: B-Leuk <4x109/l, ei-korkean riskin kromosomilöydös ja normaali LD
- harkitaan, jos atsasitidiinihoidolle ei saada vastetta (korkean riskin kromosomilöydöspotilaat eivät kuitenkaan hyödy)
- voidaan käyttää ennen allogeenista kantasolujensiirtoa pienentämään ytimen blastiosuutta
- pitkittyneen luuydinlaman vaara, hoidon mortaliteetti 10-20%
Hoitovaste
- CR 40-60%, kesto 10-12 kk
- 10%:lle pitkäaikainen CR (nuoria potilaita, joilla normaali karyotyyppi)
- ei elinaikahyötyä, ellei päästä CR-tilanteeseen ja sen jälkeen allogeeniseen kantasolujensiirtoon
2.3 Matala-annoksinen solunsalpaaja
- harkitaan allogeenisen kantasolujensiirron ulkopuolella oleville korkeamman riskin taudissa, jos atsasitidiiniä tai monisolunsalpaajahoitoa ei voi käyttää
- ei vaikutusta elinaikaan tai leukemisoitumisaikaan
- vasteet lyhyitä
Matala-annoksinen Ara-C
- annos 20mg/m2/pv sc 14-21 pv, kuukauden välein
- hoitovasteet: CR 15-20%, PR 15-20% (korkean riskin kromosomilöydös huonontaa vastetta)
- ei elinaikaetua
2.4 Allogeeninen kantasolujensiirto
Indikaatio
- <70-vuotias korkeamman riskin potilas, jolla ei ole merkittävää komorbiditeettia
- alle 50-vuotiailla tai luuytimenvajaatoimintaoireyhtymän fenotyypin tai viitteellisen sukuanamneesin yhteydessä tulisi poissulkea periytyvät MDS-alttiudet ja telomeropatiat, koska ne vaikuttavat mahdollisen sisarusluovuttajan valintaan ja siirron esihoitoon
- kantasolujensiirtokeskuksen konsultaatio
- päivitetty kantasolujensiirto-riskiluokitus (Sorror ML, Blood 2013); netissä on linkki siihen pohjautuvaan riskilaskuriin: http://www.hctci.org/
Toteutus
- nuoremmille potilaille täysimittainen esihoito
- vanhemmille potilaille tai niille, joilla on merkittäviä oheissairauksia, kevennetty esihoito, esimerkiksi treosulfaanipohjainen
- jos ytimen blastiosuus >10%, ennen siirtoa intensiivinen solunsalpaajahoito tai atsasitidiini
Hoitovaste
- keskimäärin 35-40% pitkäaikaiseen remissioon
- sytogenetiikka ja ytimen blastiosuus tärkeimmät relapsiriskiä ennustavat tekijät
- pitkäaikaistulokset (OS) selvästi paremmat matalamman riskin MDS:ssa (IPSS-Low 75%, IPSS-Int-1 60%) kuin korkeamman riskin taudissa (IPSS-Int-2 45% ja IPSS-High 30%)
- kompleksisessa karyotyypissä ja sekundaarisessa MDS:ssa ennuste siirrollakin huono (10%)
- toimenpidemortaliteetti 15-25% ja relapsiriski 20-30% (relapsi yleensä kolmen vuoden sisällä)
2.5 Tutkimuksissa olevia hoitoja
2.5.1 Hypometyloivat lääkkeet
Atsasitidiini
- matalamman riskin MDS:ssa
- allogeenisen kantasolujensiirron jälkeisessä relapsissa
- ylläpitohoidossa kemoterapian jälkeen
- per os -annostelu
- tutkimuksia useissa kombinaatioissa, mm.:
- atsasitidiini + lenalidomidi
- atsasitidiini + histonideasetylaation estäjä
- atsasitidiini + anti-PD-1
Guadesitabiini, SGI-110
- korkeamman riskin taudissa
- puoliintumisaika 4,5 kertaa pitempi kuin desitabiinilla
2.5.2 Immunomoduloivat lääkkeet
Lenalidomidi
- matalamman riskin ei-5q- -MDS-potilailla
- neljäsosalla punasolusiirtotarve lakkaa
- vähemmän hoitotoksisuutta kuin 5q- -oireyhtymää hoidettaessa
- erytropoietiini + lenalidomidi: punasolujensiirtotarvevasteet lisääntyivät 25%:sta 40%:iin
- korkeamman riskin MDS:ssa
- korkea blastimäärä, trombosyytit <50 ja kompleksinen karyotyyppi ennustavat huonoa vastetta
2.5.3 Kinaasi-inhibiittorit
Rigosertibi (polo-like kinaasi- (PLK) ja PI3-kinaasi-inhibiittori)
- tutkimuksia etenkin hypometyloivan lääkkeen jälkeisessä relapsissa ja korkean riskin sytogenetiikan omaavilla potilailla
Volasertibi (polo-like kinaasi-inhibiittori)
2.5.4 TGF-ß -antagonistit
Luspatersepti
- matalamman riskin taudissa
- inhiboi Smad2/3 -signalointia ja edistää kypsien punasolujen vapautumista verenkiertoon
- punasoluvaste 40%:lla, RA-MDS:ssa (SF3B1-mutaatio+) 67%:lla
- annostelu subkutaanisina pistoksina kolmen viikon välein
Sotatersepti
- vasteet kuten luspaterseptilla
2.5.5 Solunsalpaajat
Klofarabiini ja sapasitabiini (po nukelosidianalogit)
- korkeamman riskin MDS:ssa vasteita 40-50%.lle
2.5.6 Muut
Imetelstaatti (telomeraasi-inhibiittori)
- matalamman riskin taudin anemiassa
Eksatiostaatti (GSTP-1 -inhibiittori)
- matalamman riskin taudissa
Kantasolujensiirto
Kantasolujensiirto
Allogeeninen kantasolujensiirto voi olla aiheellinen hyväkuntoisella matalamman riskin potilaalla, jolla on muuhun hoitoon reagoimaton vaikea sytopenia, huonon ennusteen kromosomilöydös tai suureneva ytimen blastiosuus ja korkeamman riskin potilaalla, jolla ei ole merkittävää komorbiditeettia.
Alaryhmissä, joissa ei ole blastiylimäärää, siirtojen tulokset ovat selvästi paremmat kuin blastiylimääräisissä alaryhmissä. Jos potilaalla on blastiylimäärä > 10%, tauti pyritään hoitamaan remissioon intensiivisellä kemoterapialla tai atsasitidiinilla ennen siirtoa.
Täysimittaisella esihoidolla toteutettavassa allogeenisessa kantasolujensiirrossa potilaan on oltava alle 61-vuotias. Kevennetyllä esihoidolla allogeenisia kantasolujensiirtoja voidaan yksilöllisesti harkiten tehdä 70 vuoden ikään saakka sekä potilaille, joilla on merkittäviä oheissairauksia.
Kirjallisuutta
- Kirjaudu tai rekisteröidy kommentoidaksesi
Kirjallisuutta
Linkit eurooppalaiseen ja pohjoismaiseen ohjeeseen
https://www.ncbi.nlm.nih.gov/pubmed/23980065
Pohjoismainen ohje NMDSG:n kotisivuilla:
Yleiskatsauksia
- Myelodysplastic syndromes. Tefferi A, Vardiman JW. N Engl J Med. 2009 Nov 5;361(19):1872-85. Review.
-
Current therapeutic approaches for patients with myelodysplastic syndromes. Greenberg PL. Br J Haematol. 2010 Jul;150(2):131-43. Epub 2010 May 5. Review.
- Optimal sequencing of treatments for patients with myelodysplastic syndromes. Itzykson R, Fenaux P. Curr Opin Hematol. 2009 Mar;16(2):77-83.
- How I treat patients with myelodysplastic syndromes. Stone RM. Blood. 2009 Jun 18;113(25):6296-303.
-
Clinical management of myelodysplastic syndromes: update of SIE, SIES, GITMO practice guidelines. Santini V, Alessandrino PE, Angelucci E, Barosi G, Billio A, Di Maio M, Finelli C, Locatelli F, Marchetti M, Morra E, Musto P, Visani G, Tura S. Leuk Res. 2010 Feb 11. [Epub ahead of print]
-
Pohjoismaisen MDS-työryhmän Nordic Care Programme 2014 Guideline, Issue 6
-
Diagnosis and treatment of primary myelodysplastic syndromes in adults. Recommendations fron the European LeukemiaNet. Malcovati L, Hellström-Lindberg E, Bowen D, Adès L, Cermak J, Del Cañizo C, Della Porta MG, Fenaux P, Gattermann N, Germing U, Jansen JH, Mittelman M, Mufti G, Platzbecker U, Sanz GF, Selleslag D, Skov-Holm M, Stauder R, Symeonidis A, van de Loosdrecht AA, de Witte T, Cazzola M. Blood. 2013;122(17):2943-64.
-
Guidelines for the diagnosis and management of adult myelodysplastic syndromes. Killick SB, Carter C, Culligan D, Dalley C, Das-Gupta E, Drummond M, Enright H, Jones GL, Kell J, Mills J, Mufti G, Parker J, Raj K, Sternberg A, Vyas P, Bowen D; British Committee for Standards in Haematology. Br J Haematol. 2014 Feb;164(4):503-25
-
Myelodysplastinen oireyhtymä - monikasvoinen luuytimen toimintahäiriö. Kauppila M, Ebeling F, Kuittinen T, Pelliniemi T-T, Poikonen E, Sankelo M, Tienhaara A, Siitonen T. Suom Lääkäril 2014;35:2099-104 (http://www.fimnet.fi/cgi-cug/brs/artikkeli.cgi?docn=000041618)
-
The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Arber DA, Orazi A, Hasserjian R, et al. Blood 2016;127:2391-405. (http://www.ncbi.nlm.nih.gov/pubmed/27069254)
Metylaation vähentämishoidot
-
Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, Byrd J, Backstrom J, Zimmerman L, McKenzie D, Beach C, Silverman LR; International Vidaza High-Risk MDS Survival Study Group. Lancet Oncol. 2009 Mar;10(3):223-32. Epub 2009 Feb 21.
- Practical recommendations for hypomethylating agent therapy of patients with myelodysplastic syndromes. Steensma DP, Stone RM. Hematol Oncol Clin North Am. 2010 Apr;24(2):389-406.
- Novel approaches for myelodysplastic syndromes: beyond hypomethylating agents. Loaiza-Bonilla A, Gore SD, Carraway HE. Curr Opin Hematol. 2010 Mar;17(2):104-9. Review.
Rautakuorma, kelaatio
- Impact of iron overload in myelodysplastic syndromes. Fenaux P, Rose C. Blood Rev. 2009 Dec;23 Suppl 1:S15-9. Review.
- Iron chelation therapy in myelodysplastic syndrome - Cui bono? Tefferi A, Stone RM. Leukemia. 2009 Aug;23(8):1373.
Immunosuppressiiviset hoidot
- Cyclosporin A therapy in hypoplastic MDS patients and certain refractory anaemias without hypoplastic bone marrow. Jonásova A, Neuwirtová R, Cermák J, Vozobulová V, Mociková K, Sisková M, Hochová I. Br J Haematol. 1998 Feb;100(2):304-9
- Antithymocyte globulin for patients with myelodysplastic syndrome. Molldrem JJ, Caples M, Mavroudis D, Plante M, Young NS, Barrett AJ. Br J Haematol. 1997 Dec;99(3):699-705.
-
Antithymocyte globulin for treatment of the bone marrow failure associated with myelodysplastic syndromes. Molldrem JJ, Leifer E, Bahceci E, Saunthararajah Y, Rivera M, Dunbar C, Liu J, Nakamura R, Young NS, Barrett AJ. Ann Intern Med. 2002 Aug 6;137(3):156-63.
-
Successful treatment with cyclosporin A for myelodysplastic syndrome with erythroid hypoplasia associated with T-cell receptor gene rearrangements. Shimamoto T, Iguchi T, Ando K, Katagiri T, Tauchi T, Ito Y, Yaguchi M, Miyazawa K, Kimura Y, Masuda M, Mizoguchi H, Ohyashiki K. Br J Haematol. 2001 Aug;114(2):358-61.
-
Phase II study of rabbit anti-thymocyte globulin, cyclosporine and granulocyte colony-stimulating factor in patients with aplastic anemia and myelodysplastic syndrome. Garg R, Faderl S, Garcia-Manero G, Cortes J, Koller C, Huang X, York S, Pierce S, Brandt M, Beran M, Borthakur G, Kantarjian H, Ravandi F. Leukemia. 2009 Jul;23(7):1297-302.
Lenalidomidi
- List et al. NEJM
-
Practical recommendations on the use of lenalidomide in the management of myelodysplastic syndromes. Giagounidis A, Fenaux P, Mufti GJ, Muus P, Platzbecker U, Sanz G, Cripe L, Von Lilienfeld-Toal M, Wells RA. Ann Hematol. 2008 May;87(5):345-52.
-
Lenalidomide on alternative days is effective in myelodysplastic syndrome with 5q- deletion. Defina M, Rondoni M, Gozzetti A, Aprile L, Chitarrelli I, Fabbri A, Lauria F, Bocchia M. Br J Haematol. 2010 Feb;148(3):483-4.
TPO-agonistit
- Thrombocytopenia in Patients With Myelodysplastic Syndromes. Jeffrey Bryan, Elias Jabbour, Hillary Prescott, Hagop Kantarjian. Semin Hematol 2010;47(3):274-280.
Muut hoidot
-
Combination of 5-azacytidine and thalidomide for the treatment of myelodysplastic syndromes and acute myeloid leukemia. Raza A, Mehdi M, Mumtaz M, Ali F, Lascher S, Galili N. Cancer. 2008 Oct 1;113(7):1596-604.
Potilasohje
- Kirjaudu tai rekisteröidy kommentoidaksesi
Potilasohje
- MDS-opas:http://www.syopapotilaat.fi/pdf/mdsopas.pdf
- Suomen Lääkärilehdessä julkaistun MDS-artikkelin potilasversio: http://www.potilaanlaakarilehti.fi/tiedeartikkelit/mds-ndash-monikasvoinen-luuytimen-toimintahairio/#.VDOe0Cl_u4A
- Pohjoismaisen MDS-ryhmän potilasohje (erikseen matalamman ja korkeamman riskin MDS-potilaille) suomeksi käännettynä: http://www.nmds.org
- Alkuinformaatiovideo vastikään sairastuneelle MDS-potilaalle http://minullatodettiin.fi/index.php/work/mds/
MDS-MPN
MDS-MPN
WHO-tautiluokituksessa omana ryhmänään ovat taudit, joissa on piirteitä sekä myeloproliferatiivisista että myelodysplastisisita syndroomista. Yleisin tähän ryhmään kuuluvista taudeista on krooninen myelomonosyyttileukemia (KMML). Muita ryhmään kuuluvia tauteja ovat atyyppinen krooninen myeloinen leukemia (ICD-O 9876), juveniili myelomonosyyttileukemia (ICD-O 9946) ja luokittelematon myelodysplastinen/myeloproliferatiivinen tauti (ICD-O 9975).
| Kirjoittajat: | Mika Kontro, Kimmo Porkka |
| Viimeisin päivitys: | 21.8.2014 |
| © Suomen Hematologiyhdistys | Suomen Leukemiaryhmä |
KMML
KMML
Luokitus: ICD-O 9945/3 ICD-10 D46.7
Krooninen myelomonosyyttileukemia (KMML) on klonaalinen hematopoieettinen maligniteetti, jossa on piirteitä sekä myeloproliferatiivisesta sairaudesta että myelodysplastisesta syndroomasta. Se luokitellaan WHO 2016-revisioluokituksessa myelodysplastis-myeloproliferatiivisiin neoplasioihin, joissa KMML on yleisin alaryhmä. Sille on luonteenomaista perifeerisen veren monosytoosi ja taipumus AML-progressioon. Tautisolut on GM-CSF-hypersensitiivisiä.
KMML on iäkkäiden ihmisten tauti, mediaani-ikä diagnoosihetkellä on 71-74 vuotta. Sairaus on yleisempi miehillä (1,5-3:1). Luotettavia arvioita taudin esiintyvyydestä ei ole. Ilmaantuvuudeksi on arvioitu 0,5-1,0 uutta tapausta 100 000 henkilöä kohden vuodessa, noin viidesosa MDS:n ilmaantuvuudesta.
Noin 30%:lla potilaista todetaan sytogeneettinen klonaalinen kromosomipoikkeavuus. Eksomisekvensoinnilla voidaan osoittaa mutaatioita epigeneettisisissä säätelijäproteiineissa (mm. ASXL1, TET2, DNMT3A, IDH1-2), spliseosomissa (mm. SFB1), tuumorisupressoreissa (TP53) tai transkriptiotekijöissä/-säätelijöissä (mm. KRAS, NRAS, FLT3, RUNX1, JAK2) noin 90% potilaista.
Valtaosalla potilaista todetaan diagnoosivaiheessa leukosytoosi ja näillä potilailla myeloproliferatiiviset piirteet ovat selkeimmät. Osalla potilaista leukosyyttiluku on normaali tai matala, ja näillä potilailla puolestaan taudinkuva muistuttaa myelodysplastista syndroomaa.
Koska monosyyteillä on taipumus hakeutua kudoksiin, todetaan kroonisessa myelomonosyyttileukemiassa usein ekstramedullaarisia ilmentymiä. Ienmuutokset ovat tavallisia ja potilailla voidaan todeta makulopapulaarisia ihomuutoksia. Myös pleuraaliset ja perikardiaaliset nestekerymät sekä askites ovat mahdollisia. Tavallisimmin oireet ovat sytopenioiden tai myeloproliferaation (väsymys, hikoilu, laihtuminen) aiheuttamia.
Krooninen myelomonosyyttileukemia muuttuu akuutiksi leukemiaksi noin 20%:lla potilaista. Potilaiden keskimääräinen elinaika on 20-40 kuukautta.
Pohjoismaissa meneillään olevia tutkimuksia
Pohjoismainen MDS-ryhmä:
1) NMDSG14A: a phase I/II, Open-Label, Multicenter Study of the Safety, Efficacy and Immune Response of Histamine Dihydrochloride and Low-dose IL-2 in CMML
2) NMDSG14B: Individual molecular minimal residual disease monitoring for patients with MDS and mixed MDS/MPN treated with allogeneic stem cell transplantation
3) pohjoismainen retrospektiivinen tutkimus KMML-allosiirroista aikavälillä 1/08-12/15, Lone Smidstrup Friis
HUS, hematologian klinikka:
MDS-MOLE-2015: Lääkehoidon aikainen mutaatioseuranta, allosiirron jälkeinen kvantitatiivinen mutaatiomäärän MRD-seuranta, yhteyshenkilöt Freja Ebeling, Johanna Illman
| Kirjoittajat: | Freja Ebeling, Mika Kontro |
| Viimeisin päivitys: | 03.02.2017 |
| © Suomen Hematologiyhdistys |
Diagnoosi
Diagnoosi
Diagnostiset kriteerit WHO:n mukaan (2008-luokitus + 2016 revisio)
- Persistoiva (vähintään 3 kk) veren monosytoosi vähintään tasoa 1 x 109/l ja monosyyttejä erittelyjakaumassa vähintään 10% leukosyyteistä
- Ei täytä BCR-ABL1-pos KML:n, primaarisen myelofibroosin, PV:n tai ET:n WHO-diagnoosikriteerejä
- Ei PDGFRA-, PDGFRB- tai FGFR1-uudelleenjärjestymää tai PCM1-JAK2-fuusiogeeniä, jos potilaalla on eosinofiliaa
- Vähemmän kuin 20% blasteja (mukaanlukien myeloblastit, monoblastit ja promonosyytit) veressä ja luuytimessä
- Dysplasia yhdessä tai useammassa solulinjassa. Jos dysplasiaa ei ole, voidaan KMML-diagnoosi kuitenkin tehdä, mikäli muut kriteerit täyttyvät ja (1) on osoitettavissa muu hankinnainen klonaalinen syto- tai molekyyligeneettinen poikkeavuus hematopoieettisessa solukossa (etenkin KMML:ssä usein nähdyissä mutaatioissa TET2-, SRSF2-, ASXL1- ja SETBP1 -geeneissä) tai (2) monosytoosi on jatkunut vähintään kolme kuukautta ja (3) kaikki muut monosytoosin syyt ovat poissuljettuja
Oireet ja löydökset
- Yleisoireina laihtumista, yöhikoilua, lämpöilyä ja väsymystä
- Ihoaffisiossa makulopapulaarinen ihottuma
- Ienhyperplasia osalla potilaista
- Splenomegalia (50%), hepatomegalia (20%)
- N. 20%:lla immunologisia ilmentymiä, kuten perikardium- tai pleuraeffuusio, joskus askites, vaskuliitteja, tyreoidiitti, ITP. Myös keuhkomuutoksia on kuvattu.
- Lymfadenopatiaa ei yleensä todeta. Mahdollinen lymfadenopatia voi viitata transformaatioon.
- Perussairauksiin liittyvät löydökset (komorbiditeetti/hoitovalinnat)
Luuydinnäyte
- Luuydinaspiraatin morfologinen tutkimus rautavärjäyksineen
- Luuydinbiopsia (Bm-PAD)
- G-raitatutkimus ja BCR/ABL-fuusiogeenitutkimus Ph-kromosomin poissulkemiseksi, eosinofilian yhteydessä PDGFRA-, PDGFRB- ja FGFR1-uudelleenjärjestymä- ja PCM-JAK2-fuusiogeenitutkimus
- Tarvittaessa luuytimen virtaussytometrinen tutkimukset
- Myelooisten mutaatioiden seulontatutkimus harkinnan ja saatavuuden mukaan
Veren tai luuytimen monosyyttien immunofenotyyppi ja karyo- tai genotyyppi
- KMML-solujen immunofenotyyppi: CD13+, CD33+, CD14±, CD64±, CD68±, CD163±
- Kromosomipoikkeavuuksia 20-40%:lla potilaista,tavallisimmat karyotyyppimuutokset: +8, -7/del(7q), rakenteelliset poikkeavuudet 12p.
- Mikäli merkittävä eosinofilia ja todetaan jokin dg-kriteerien kohdassa mainituista geenipoikkeavuuksista, ei kyseessä ole KMML, vaan tauti luokittuu omaan myelooisten/lymfaattisten neoplasioiden alaryhmäänsä WHO-luokituksessa
Kuvantamistutkimukset
- THX-rtg
- Vatsan UÄ
Erotusdiagnoosi
Erotusdiagnoosi
Monosytoosin muita syitä:
- Krooniset infektiot:
- Tuberkuloosi
- Brucelloosi
- Endokardiitti
- Kollagenoosit
- SLE
- Temporaaliarteriitti
- Nivelreuma
- Hematologiset maligniteetit
- Hodgkinin lymfooma
- AML M5
- Luuytimen toipumisessa reaktiivisena muutoksena
- G-GSF
- Malaria
Seuranta ja vastearvio
Seuranta ja vastearvio
Seuranta
Oireetonta potilasta, jonka tauti on stabiili voidaan seurata hoidoitta. Iäkkäitä potilaita, joiden muut sairaudet rajoittavat hoitoa, voidaan seurata terveyskeskuksessa tai aluesairaalassa. Mikäli ikä ja yleiskunto mahdollistavat aktiivisen hoidon, tulee hoitovastetta seurata mahdollisuuksien mukaisesti esimerkiksi molekyyligeneettisin menetelmin. Hoidossa olevia potilaita seurataan tapauskohtaisesti.
Hoitovasteen arviointi
Diagnoosivaiheessa tehtyjen tutkimusten perusteella valitaan potilaskohtaiset seurantatutkimukset (morfologia, sytogenetiikka, molekyyligenetiikka, immunofenotyypitys), mikäli sellaisia on olemassa. Joka vastearviokerralla tulisi määrittää seurannan avainmuuttujat sekä määrittää taudin status.
Taudin status
Taudin status
Vastearvion yhteydessä pyritään tautitaakan arvio ja taudin tilanne luokittelemaan oheisen taulukon mukaisesti (SHR-muuttuja: status-mds-mpd). Taulukosta valitaan arviointihetkellä oleva paras vaste.
|
Koodi |
Lyhenne |
Nimike |
Määritelmä |
|
0 |
Dg |
Diagnoosivaihe |
|
|
1 |
CR-MRDneg |
Remissio, jäännostauti negatiivinen |
|
|
2 |
CR-MRDpos |
Remissio, jäännostauti positiivinen |
|
|
3 |
CCR |
Sytogeneettinen remissio (complete cytogenetic response) |
|
|
4 |
pCR |
Osittainen sytogeneettinen vaste (partial cytogenetic response) |
|
|
5 |
CR |
Morfologinen remissio (complete remission) |
|
|
6 |
PR |
Osittainen vaste (partial remission) |
(esimerkki: blastiosuus 15%-> 6%)
|
|
7 |
HI |
Verenkuva-arvojen korjaantuminen (hematologic improvement) |
(esim: B-neut 0,3-> 0,6) |
|
8 |
Stabiili |
Stabiili tauti (stable disease) |
|
|
9 |
Progressio |
Taudin eteneminen (progressive disease) |
|
|
10 |
Relapsi |
Relapsi täydellisestä tai osittaisesta vasteesta |
|
|
90 |
Exitus-relapsi |
Exitus, progressio |
|
|
91 |
Exitus-infektio |
Exitus, infektio |
|
|
92 |
Exitus-other |
Exitus, muu syy |
|
|
99 |
Exitus-tuntematon |
Exitus, syy tuntematon |
|
|
999 |
Poistunut seurannasta |
Lost to follow-up |
|
Luokittelu ja ennuste
Luokittelu ja ennuste
Luokittelu WHO-luokituksen 2016 -revision mukaan
| KMML-0 |
<2% blasteja veressä ja <5% blasteja luuytimessä |
|
KMML-1 |
2-4% blasteja veressä ja/tai 5-9% blasteja luuytimessä |
|
KMML-2 |
5-19% blasteja veressä tai 10-19% blasteja luuytimessä tai mikäli todetaan Auerin sauvoja (blasti-/promonosyyttimäärästä riippumatta) |
KMML:n ilmentymien perusteella taudin fenotyyppi jaetaan kahteen päätyyppiin: dysplastinen "MDS-KMML" ja myeloproliferatiivinen "MPN-KMML" (FAB-KMML-alatyypit). Ensinmainitulle luonteenomaista ovat sytopeniat ja jälkimmäiselle Leuk-taso >13x106/l, hepato- ja splenomegalia, ekstramedullaariset ilmentymät, yleisoireet ja huonompi ennuste.
Ennuste
KMML:n ennusteen arvioimiseksi on kehitetty lukuisia riskiluokituksia. MDS:n kaltaista vakiintuneita riskiluokitusta ei ole. Alla esitelty kaksi erilaista riskiluokitusta ennusteen arvioimiseksi. Tulevaisuudessa mutaatiotieto vaikiintunee osaksi ennustearviota.
Mayo prognostic model (Patnaik et al, Leukemia 2013)
|
Monosyyttiluku >10x109/l |
1 piste |
|
Blasteja veressä |
1 piste |
|
Hemoglobiini <100 g/l |
1 piste |
|
Trombosyytit <100 x109/l |
1 piste |
Riskin mukainen elinaikaennuste Mayo-mallissa
|
0 pistettä |
32kk |
|
1 pistettä |
18 kk |
|
≥2 pistettä |
10 kk |
CPSS-riskiluokitus (Such et al, Haematogica 2011, Such et al. Blood 2013;121:3005-15)
Neljä muuttujaa, joilla OS- ja LFS-ennustearvo:
- FAB-KMML-alatyyppi, Leuk <13 vs ≥ 13 (1 piste)
- WHO-KMML-alatyyppi, KMML-1 vs KMML-2 (1 piste)
- Punasolusiirtoriippuvuus (1 piste)
- sytogeneettinen riskiluokka, espanjalainen luokitus (max 2 pistettä)
- low (normal, -Y), intermediate (muut), high (+8, kompleksi, krom 7 poikk)
| Riskiluokka | Pisteitä | Median OS (kk) |
|
Low |
0 |
72 |
|
Intermediate-1 |
1 |
31 |
|
Intermediate-2 |
2-3 |
13 |
| High | 4-5 | 5 |
CPSS-Molecular model (Elena C. et al. Blood 2016;128:1408-17, pisteytys: Table 2 ja 3, elossaolokäyrät Figure 3)
- tässä mutaatiostatuskin mukana (ASXL1, NRAS, RUNX1, SETBP1)
Hoito
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hoito
Krooninen myelomonosyyttileukemia on heterogeeninen tautiryhmä. Osalla potilaista luuytimen toiminta hiipuu ja hoitoa vaativia sytopenioita kehittyy. Toiset potilaat puolestaan tarvitsevat sytoreduktiivista hoitoa joskus huomattavankin leukosytoosin vuoksi. Potilaat ovat valtaosin iäkkäitä, ja muut sairaudet saattavat rajoittaa hoidon valintaa. Näiden potilaiden hoito onkin valtaosin oireita lievittävää. Tutkimustietoa KMML:n hoidosta on vähäisesti. Yhteneviä hoitosuosituksia ei ole. MDS:n hoitoon kehitettyjä hypometyloivia lääkeaineita on käytetty myös KMML-potilaiden hoitoon ja osa hoitotuloksista on lupaavia. Allogeenisen kantasolusiirron mahdollisuus kartoitetaan hoitoon soveltuvilla potilailla.
Oireettomia potilaita, joiden tauti on stabiili, seurataan. Infektioihin voi liittyä huomattava leukosytoosi, joka ei useinkaan vaadi muuta hoitoa
Tukihoidot
- Anemia: punasolujensiirrot; potilas voi hyötyä EPO:sta, rautakelaatiota ei toistaiseksi suositella
- Trombosytopenia: trombosyyttisiirrot tarvittaessa.
- Neutropenia: G-CSF neutropenisissa infektioissa. Voidaan harkita toistuvien infektoiden estoon. Splenomegalian yhteydessä profylaktiseen käyttöön liittynee lisääntynyt pernaruptuurariski.
Sytoreduktiivinen hoito
Hydroksiurea
- Soveltuu myeloproliferatiivisen tautimuodon hoitoon.
- Tehoaa leukosytoosiin, ihoinfiltraatteihin, onteloiden nestekertymiin sekä mahdollisesti splenomegaliaan.
- OR 60% (Wattel et al. 1996)
- Hyrdoksiurean käyttöä tulisi välttää atsasitidiinin aikana mahdollisen antagonismin vuoksi.
Hypometyloivat lääkeaineet
5-atsasitidiini (Vidaza®)
- soveltuu dysplastisen tautimuodon hoitoon
- 75 mg/m2/vrk sc 7 pv kuurein, syklin pituus 28 vrk
- Useimmiten käytetään annostelua 75mg/m2/vrk päiväaikataululla 5-0-2 (=viikonlopputauko). Vaihtoehtoisten annostelutapojen tehosta ei ole vertailua vakioannostukseen verrattuna.
- haittavaikutuksia eniten kahden ensimmäisen syklin aikana:
- myelosuppressio, pahoinvointi, GI-oireet, injektiokohdan ärsytys
- Vaste saavutetaan hitaasti, luuytimen vastearviota suositellaan aikaisintaan neljän syklin jälkeen.
- OR 30-40%, CR n. 15%, OS 12-37 kk
- Atsasitidiinin tehosta ei ole vertailevia tutkimuksia KMML-potilailla (esim. tukihoitoon tai pieniannos-sytarabiiniin verrattuna).
Allogeeninen kantasolujensiirto
- ainoa kuratiivinen hoitomuoto, korkea hoitoon liittyvä kuolleisuus
- ei prospektiivisia tutkimuksia
- EBMT, 283 potilaan aineisto: OS 42% (Symeonidis et al. 2010)
- nuoremmat korkeamman riskin potilaat, joilla ei estävää komorbiditeettia
- jos KMML-2 tai RIC-esihoito, harkitaan sytoreduktiivista tai demetyloivaa hoitoa ennen siirtoa
- Allogeenista kantasolujensiirtoa puoltavat suuren riskin tauti, epäsuotuisan ennusteen sytogenetiikka, blastiylimäärä tai tankkaustarve.
- Taulukko: Mayo-klinikan modifioitu algoritmi lääkehoidon valintaan fenotyypin ja blastiosuuden mukaan. Laaja-alaisesti hyväksyttyjä, yhtenäisiä hoitosuosituksia KMML:aan ei ole. (Patnaik, Tefferi et al, Br J Haematol, 2014)
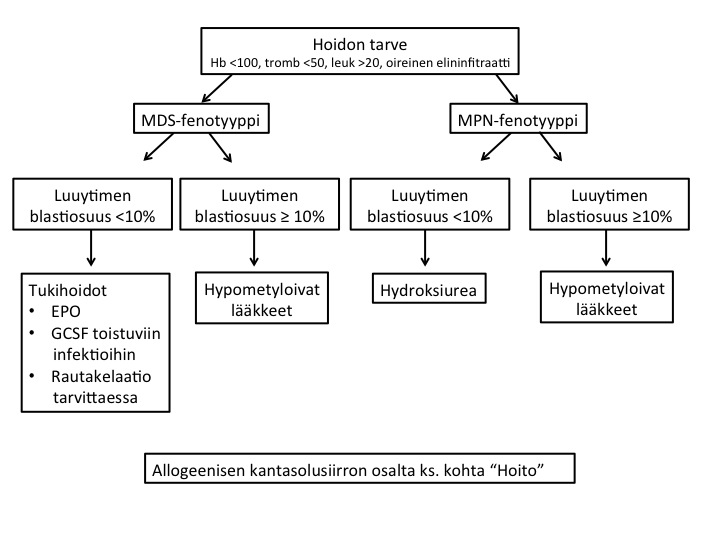
Tulevaisuuden mahdollisia hoitoja
Sotatersepti (ACE-011), luspatersepti (ACE-536)
Eltrombopagi
Ruksolitinibi proliferatiivisessa KMML:ssa
anti-GM-CSF-vasta-aineet
IDH-estäjät IDH-mutatoituneille
Farnesyylitransferaasiestäjät
Kirjallisuutta
Kirjallisuutta
- Patnaik MM, Parikh SA, Hanson CA, Tefferi A. Chronic myelomonocytic leukemia: a concise clinical and pathophysiological review. British Journal of Haematology, 2014, 165, 273-86
- Emanuel PD. Juvenile myelomonocytic leukemia and chronic myelomonocytic leukemia. Leukemia 2008;22, 1335-42
- Chen W, Huan Q. Detection of FLT3/ITD, JAK2(V617F) and NMP1 gene mutations in chronic myelomonocytic leukemia. Leukemia Research 33 2009 e207-e209
- Onida M, Kantarjian HM, Smith TL et al. Prognostic factors and scoring system in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood 2002;99:1, 840-9
- Oninda F, Beran M. Diagnosis and management of chronic myelomonocytic leukemia. Current hematologic malignancy reports 2008, 3:31-36
- Reiter A, Invernizzi R, Cross NCP et al. Molecular basis of myelodysplastic/myeloproliferative neoplasms. Haematologica, Vol 94, Issue 12, 1634-1638
- Such E et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Hematologica 2011, 96, 375-83
- Orazi A, Bennett J.M, Germin U et al. Chronic myelomonocytic leukemia. Kirjassa: WHO Tumors of hematopoietic and lymphoid tissues. 2008
- Patnaik MM et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia 2014, epub ahead of print.
- Siitonen T, Koistinen P. Myelodysplastiset oireyhtymät. Kirjassa: Veritaudit 2007.
Myeloproliferatiiviset taudit
- Kirjaudu tai rekisteröidy kommentoidaksesi
Myeloproliferatiiviset taudit
Polysytemia vera
Polysytemia vera
(lyhyt yhteenveto)
| Kirjoittajat: | Ulla Wartiovaara-Kautto, Riitta Niittyvuopio SLR:n MPD-ryhmä |
| Viimeisin päivitys: | 14.06.2010 |
| © Suomen Hematologiyhdistys | Suomen Leukemiaryhmä |
Diagnoosi
Diagnoosi
Polysytemia veran diagnoosi perustuu tyypilliseen verenkuvaan (erytrosytoosi), veressä todettuu JAK2-geenin mutaatioon ja sekundaaristen syiden poissulkuun. Ks. tarkemmin WHO:n diagnostisit kriteerit (2008).
A. Diagnoosivaiheen ensilinjan tutkimukset
- Verikokeet: B -PVK+TKD, P-Ferrit, fP-Fe, fP-Trfesat, P-CRP, B-La, P-Krea, P-LD, P-Urea, P-Uraat, P-K, P-Na, P-ASAT, P-AFOS, P-Bil, P-Bil-Kj, P-Gluc, fP-Kol-LDL (osatutkimukset fP-Kol, fP-Kol-HDL ja fP-Trigly), B-JAK2-D
- Saturaatio pulssioksimetrillä (sairaskertomusmerkintä)
- Luuydinaspiraatio (morfologia ja rautavärjäys), luuydinbiopsia
- Hematologinen kromosomitutkimus (Bm-KromHem)
- Kantasoluviljely (luuydin tai veri); ei pakollinen tutkimus
- Kuvantamistutkimukset: thorax, vatsan uä
- Ekg
B. Diagnoosivaiheen toisen linjan tutkimukset, jos erytrosytoosin syy ei edellä olevilla tutkimuksilla varmistu luotettavasti
- Verikokeet: B-Hb-CO, aB-Astrup, S-EPO, B-Hb-Fr, Pt-EryVoIN (punasolutilavuuden määritys)
- Kuvantaminen: vartalon TT
C. Erotusdiagnoosi
WHO:n kriteerit
WHO:n kriteerit
Pääkriteerit
- Hemoglobiini > 185 g/L miehillä ja >165 g/L naisilla, tai muu osoitus suurentuneesta punasolumassasta
- JAK2V617F-mutaatio
Sivukriteerit
- Luuydinbiopsiassa myelooisten linjojen (erytrooinen, granulosyytti-makrofagi ja megakaryosyytti) proliferaation aiheuttama kokonaissolukkuuden lisääntyminen
- Seerumin erytropoietiinin pitoisuus viitealuetta pienempi
- Erytrooinen spontaanikasvu kantasoluviljelyissä
Diagnoosi edellyttää molempien pääkriteerien ja yhden sivukriteerin tai yhden pääkriteerin ja 2 sivukriteerin täyttymistä.
P. veran aiemmat diagnostiset kriteerit (WHO 2001)
- A1. Punasolumassa koholla (> 25 % yli normaalialueen), tai miehillä Hb > 185 g/L, naisilla Hb >165 g/L
- A2. Ei viitettä sekundaarisesta erytrosytoosista
familiaalinen erytrosytoosi poissuljettuei EPO-tason nousua, joka johtuisi hypoksiasta (saturaatio < 92%)hapen sitomiskyvyltään poikkeavasta hemoglobiinista. EPO-reseptorin poikkeavuudestatuumorin epätarkoituksenmukaisesta EPO-tuotannosta - A3. Splenomegalia
- A4. Klonaalinen kromosomimuutos (ei Ph-kromosomi tai BCR/ABL fuusiogeeni)
- A5. Erytrooinen spontaanikasvu in vitro – viljelyssä
- B1. B-tromb > 400 x 109 /L
- B2. B-leuk > 12 x 109 /L
- B3. Luuydinbiopsiassa kokonaissolukkuus lisääntynyt, erytrooisen ja megakaryosyyttisarjan proliferaatio
- B4. Matala EPO-taso
PV voidaan diagnosoida, mikäli A1 + A2 ja jokin muu A-kriteeri täyttyy, tai A1 + A2 ja kaksi B-kriteeriä täyttyy.
Erotusdiagnoosi
Erotusdiagnoosi
Erytrosytoosin syyt
| A. Relatiivinen erytrosytoosi | Stressi, kuivuminen, diureetit, palovammat | |
|
B. Absoluuttinen erytrosytoosi |
1. Polysytemia vera | |
| 2. Sekundaariset erytrosytoosit | a. Huono hapenkuljetuskapasiteetti: - Hengitysvajaus: COPD, obesiteetti, Pickwickin syndrooma, vuoristo, ym. - Kardiovaskulaariset sairaudet: syanoottiset synnynnäiset sydänviat, septumdefekti ym. - Hemoglobiinin funktiohäiriöt: hemoglobinopatiat, runsas tupakointi |
|
|
b. Epätarkoituksenmukainen erytropoietiinin eritys: |
||
PNH
- Kirjaudu tai rekisteröidy kommentoidaksesi
PNH
Tämä kansallinen suositus on hematologian erikoislääkärien (PNH-ryhmän) laatima kannanotto PNH-taudin diagnostiikasta, hoidosta ja seurannasta Suomessa.
Suomen PNH-ryhmä
Elina Armstrong (pj), Hanna Jarva, Eva-Stina Korkama (siht), Minna Lehto, Seppo Meri, Tarja-Terttu Pelliniemi, Eira Poikonen, Jonna Salonen, Marjatta Sinisalo sekä Marjaana Säily
Johdanto
PNH-tauti, kohtauksittainen yöllinen hemoglobiinivirtsaisuus, on hankittu oireyhtymä, jonka kliinisiin pääpiirteisiin kuuluvat suonensisäinen hemolyysi, laskimotukokset sekä luuytimen toimintahäiriö (1). Kyseessä on hyvin harvinainen sairaus: vuotuisen ilmaantuvuuden arvioidaan olevan 1,3/miljoonaa asukasta kohden (2). Suomessa diagnosoidaan arviolta muutama potilas vuosittain.
Koska kyseessä on hyvin harvinainen tauti, on tärkeää luoda yhtenäiset kansalliset hoitolinjat. Suosituksia PNH-taudin hoidosta on tehty mm Ruotsissa, Norjassa sekä Australiassa. Tämä kansallinen suositus on hematologian erikoislääkärien (PNH-ryhmän) laatima kannanotto PNH-taudin diagnostiikasta, hoidosta ja seurannasta Suomessa. Suositus pyritään päivittämään parin vuoden välein.
PNH-taudin hoitoon on vuodesta 2007 lähtien ollut käytössä monoklonaalinen vasta-aine, ekulitsumabi. Kyseessä on huomattavan kallis hoitomuoto, minkä vuoksi hoitoindikaatio on tarkkaan harkittava. PNH-ryhmää voi konsultoida potilastapauksista ja ennen ekulitsumabihoidon aloitusta.
Patofysiologia
Potilaiden oireet johtuvat komplementin aktivaatiosta solukalvoilla (3). PNH-potilaiden verisoluista puuttuu kaksi komplementin tärkeää suojamolekyyliä, protektiini (CD59) ja DAF (decay accelerating factor, CD55), joiden tehtävänä on estää komplementin aktivaatiota elimistön omien solujen pinnalla (4-6). CD59 ja CD55 ovat normaalisti kiinnittyneinä solukalvoon glykofosfoinositoli (GPI) ankkurilla. PNH-potilailla on mutaatio ankkurimolekyylin synteesiin tarvittavaa entsyymiä koodaavassa geenissä (PIG-A-geeni) ja sen seurauksena GPI-ankkuri puuttuu solujen pinnasta (7).
Diagnostiikka
Diagnostiikka
PNH on kliininen diagnoosi, joka tulee varmistaa osoittamalla GPI-ankkurimolekyylin puutos neutrofiilien, monosyyttien ja punasolujen virtaussytometrisella tutkimuksella. PNH-klooni voidaan todeta, jos GPI-ankkurimolekyylin puutos on osoitettavissa vähintään kahdessa eri solulinjassa. Koska PNH-kloonin koko määräytyy neutrofiilien kloonikoon perusteella, on suositeltavaa tutkia ensi vaiheessa neutrofiilien ja monosyyttien GPI-ankkurimolekyylin puute. GPI-puutoksen osoittamiseen käytettävät vasta-aineet / väriaineet ovat vähintään seuraavat: neutrofiileista anti-CD24 ja FLAER (fluoresiinilla leimattu proaerolysiini) , monosyyteistä anti-CD14 ja FLAER, punasoluista anti-CD59. GPI-puutteisten solujen osuus ilmoitetaan prosentteina tutkittavasta solupopulaatiosta. Punasoluista ilmoitetaan erikseen tyyppi III:n solut (täydellinen GPI-puutos) ja tyyppi II:n solut (osittainen GPI-puutos).
Laboratoriokohtaiset tutkimusnimikkeet ja lähetysohjeet tulee tarkistaa ennen näytteenottoa. Tuloksista annetaan lausunto.
Esimerkki löydösten tulkinnasta:
- PNH-klooni voidaan todeta jos potilaalla vähintään kahdessa solulinjassa >1% GPI-puutteisia soluja. Kliinisten löydösten, luuydintutkimusten ja hemolyysimarkkeritutkimusten perusteella voidaan todeta onko kyseessä 1) Klassinen PNH 2) PNH liittyneenä klonaaliseen verisairauteen tai 3) Subkliininen PNH.
- Seurattava tilanne (pieni PNH-klooni), jos GPI-puutteisia soluja on 0.1 – 1%
- Yksittäisiä GPI-puutteisia soluja, jos niitä on alle 0.1%
- Jos GPI-puutteisia soluja ei löydy, ilmoitetaan tutkimuksen herkkyys (25 / analysoitujen neutrofiilien määrä x100%).
Indikaatioita PNH-kloonin tutkimiselle:
- Coombs negatiivinen hemolyyttinen anemia
- hemoglobinuria, tummavirtsaisuus
- aplastinen anemia
- pienen riskin MDS
- hematologin toteama epäselvä sytopenia
- tromboosi epätavallisessa paikassa, mm. vatsan alueella, vaikeahoitoinen,
tukostaipumustutkimuksissa (P-Trombot) ei selittävää, maligniteettiselvittelyt tehty
Muut tutkimukset harkinnan ja tarpeen mukaan:
- Luuytimen morfologia, tarvittaessa kromosomitutkimukset ja biopsia (potilailta, joilla pansytopenia)
- TVK, retik, CRP, eGFR,, ALAT, AFOS, bil, LD, haptoglobiini, Mg, alb-Ca, pro-BNP, lipidit, gluk, transferriinisaturaatio,TfR, ferritiini ,TSH, T4v, hepatiittiserologia, HIV-va, U-kemseu, EPO, B12-TC2, E-folaatti
- Hyytymistutkimukset (APTT, TT, Trombai, Fibr ja FiDD) ja tromboositaipumustutkimukset, P-Trombot; FII-D, FV-D, APCres, AT3, B2GPAbG, FVIII, KardAbG, LupusAK, PC, PS-AgV, TT ja Trombai)
- EKG, THX-rtg
- Vatsan ultraääni, sydämen ultraääni
Klassisessa PNH:ssa potilailla GPI-puutteisten (PNH-klooni) solujen osuus on yleensä yli 10%. Taudinkuva voi ajan kuluessa muuttua, potilaalle voi seurannan aikana kehittyä luuytimen toimintahäiriö tai alkujaan luuytimentoimintahäiriöön liittyvä PNH voi muuttua klassiseksi PNH:ksi, johon liittyy hemolyysi.
Tämän suosituksen hoito- ja seurantakappaleet koskevat klassista PNH:ta.
Diagnoosin asettamisen jälkeen kaikille PNH-potilaille annetaan diagnoosi/hoitokortti (kansallinen hoitokortti on kehitteillä) ja tieto PNH-taudista kirjataan sairaskertomusjärjestelmän riskitietoihin (esim: Kohtauksittainen yöllinen hemoglobinuria, PNH. Merkittävä verisuonitukosten ja komplikaatioiden riski etenkin altistetilanteissa. Aina PNH-poliklinikan tai hematologin konsultaatio (konsultin/päiv puh.nro)).
Hoito
Hoito
Potilaiden hoito on oireenmukaista; käytetään punasolujensiirtoja, foolihappo-, rauta- ja erytropoietiinisubstituutiota, immunosupressiivistaa hoitoa (steroidit, siklosporiini) ja antikoagulaatiota. Ainoa parantava hoito on allogeeninen kantasolujensiirto, johon PNH-potilailla liittyy kuitenkin huomattava toimenpidemortaliteetti.
Antikoagulaatiohoito:
Tukoksen sairastaneilla antikoagulaatiohoito on pysyvä. Antikoagulaatiota tulee harkita kaikille potilaille, joilla on suuri PNH-klooni (>50%), etenkin jos potilaalla on runsaasti II-tyypin punasoluja tai todettu trombofilia. Ensisijaisesti suositellaan varfariinia INR-tavoitteella 2-3 tai pienimolekyylistä hepariinia (LMWH) painonmukaisella hoitoannoksella. Aspiriinia 100mg x1 ja/tai pieniannoksista statiinia voi harkita (ei näyttöä). Toistaiseksi suoria antikoagulantteja ei suositella (ei näyttöä). Altistetilanteissa (esim. sairaalahoito, infektiot, immobilisaatio) tehokas lääkkeellinen (LMWH) profylaksi on aiheellinen.
Anemian hoito:
Punasolusiirtoja pääsääntöisesti vain oireiseen anemiaan, mielellään vain yksi yksikkö kerrallaan. Mahdollista raudankertymistä monitoroidaan ja tarvittaessa harkitaan rautakelaatiohoitoa.
Rautaa annetaan raudanpuutteessa ja foolihappoa (5mgx1) kaikille, joilla hemolyysia. Potilaat, joilla todetaan matala S-EPO taso (< 200 U/l), voivat hyötyä erytropoietiinista.
Ekulitsumabihoito:
Ekulitsumabi on PNH:n hoitoon tarkoitettu humanisoitu monoklonaalinen vasta-aine, joka sitoutuu C5-komplementtiproteiiniin ja estää komplementtikaskadin terminaalista aktivaatiota ja siten PNH-positiivisten punasolujen hajoamista. Ekulitsumabin on osoitettu tehokkaasti vähentävän hemolyysiä, anemiaa, punasolusiirtotarvetta sekä tukostapahtumia PNH-potilailla(12-14).
Ekulitsumabi-hoidon aloituksen harkitsemisen kriteerit:
- PNH-diagnoosi ja aktiivinen hemolyysi
- Lisäksi vähintään yksi seuraavista:
- Tromboosi
- Punasolusiirtotarve >6-8 yksikköä/vuosi
- Hemolyysistä johtuva anemia: oireettomalla potilaalla Hb<70, oireisella Hb<100
- Hemolyysistä tai tromboosista johtuva pulmonaalihypertensio tai keuhkojen vajaatoiminta: NYHA III
- Hemolyysistä johtuva munuaisten vajatoiminta (GFR<60ml/min)
- Sileälihasspasmi (esim nielemisvaikeus, vatsakivut, erektiohäiriöt)
Raskaus ja lapsivuodeaika (3kk)
Lääkehoidon aloituksen kontraindikaatiot (poissulkukriteerit):
- PNH-klooni <10%
- kliininen harkinta, jos tromb<30, neutr<0.5
- LD<1.5x normaalin yläraja
- hoitamaton N.meningitidis- tai muu bakteeri-infektio
- komplementin puutostila
- kantasolujensiirron jälkitila
- yliherkkyys ekulitsumabille tai sen ainesosille
- potilaan huono hoitomyöntyvyys
Ekulitsumabi annostellaan iv infuusiona: 600mg iv kerran viikossa x4, sitten viikosta 5 alkaen 900mg iv joka toinen viikko toistaiseksi.
Meningokokki-infektioiden ehkäisy:
Ekulitsumabi estää komplementin terminaalista osatekijää (C5), joka on tärkeässä roolissa neisseria-infektioiden torjunnassa (Neisseria meningitidis, Neisseria gonorrhae). Tämän vuoksi ekulitsumabia saavat potilaat on rokotettava meningokokkia vastaan.
Rokotteet annetaan ekulitsumabihoidon aloituksen yhteydessä. Tämä on tarpeen, koska immunisaatio voi aiheuttaa komplementin aktivaation ja laukaista PNH-paroksysmin lisäten komplikaatioiden, esim. tromboosin riskiä. Potilaan suojaamiseksi ennen rokotevastetta aloitetaan antibioottihoito (esim. V-penisilliiniä 1milj IU 1x2 tai penisilliniallergisille roksitromysiini 150mg 1x1) vähintään 2 viikon ajan. Rokotustodistus lähetetään lääkkeen toimittajalle.
Jatkuvaa peroraalista antibioottiprofylaksiaa koko ekulitsumabihoidon ajan käytetään joissain maissa (mm. Britanniassa) ja erityisesti silloin, jos potilas ei ole saanut rokotetta meningokokki B:tä vastaan.
Rokotteena käytetään nelivalenttista konjugoitua meningokokkirokotetta (Mencevax®, Nimenrix®, Menveo®), joka antaa suojan serotyyppejä A, C, W ja Y-vastaan. Tehosteannos uusitaan 3 vuoden välein. Lisäksi annetaan serotyyppiä B vastaan suojaava rokote (Bexsero® tai Trumenba®).
Ekulitsumabia saaville potilaille suositellaan kausi-influenssarokotetta vuosittain. Immunisaatiot suositellaan ajoitettaviksi heti ekulitsumabi-infuusion jälkeen tehokkaan komplementin eston turvaamiseksi.
Mikäli potilaalle nousee kuume ≥38C ja/tai yleistila heikkenee, hänen on syytä heti hakeutua hoitoarvioon päivystyspoliklinikalle. Potilasta hoitavan yksikön tulee tällöin konsultoida PNH-pkl:aa tai potilaan tuntevaa hematologia. Ekulitsumabihoidossa olevia potilaita neuvotaan pitämään aina mukanaan turvakorttia, jossa kerrotaan meningokokki-infektion ja muiden infektioiden lisääntyneestä riskistä.
Ekulitsumabi -hoidon jatkamisen arviointi:
Hoitoa jatketaan niin kauan kuin kliininen tila kohenee tai stabiloituu, oireet lievittyvät ja elämänlaatu paranee. Hoito lopetetaan, jos ei saada vastetta niihin oireisiin ja löydöksiin, joiden perusteella hoito aloitettiin, potilas ei ole hoitomyöntyväinen tai potilaalla todetaan vakava elinaikaa merkittävästi lyhentävä muu sairaus.
Raskaudenaikainen hoito
Raskaana olevilla PNH-potilailla on kohonnut komplikaatioriski (etenkin tukosriski) niin raskauden, synnytyksen kuin lapsivuodeaikana (15). Kansainvälisen käytännön mukaan raskaana olevat PNH-potilaat hoidetaan ekulitsumabilla toisesta trimesterista jatkaen vähintään 3kk postpartum (16).
Akuutit tilanteet PNH potilailla:
PNH-tauti voi aktivoitua kaikissa akuuteissa tilanteissa mm. infektioissa ja leikkauksissa, jolloin komplikaatioriski kasvaa. Potilasta neuvotaan ottamaan herkästi yhteyttä omaan PNH-hoitoyksikköön. Hoitavien tahojen tulee konsultoida hematologia tai PNH-pkl:aa.
- Jos potilas joutuu sairaalahoitoon mistä tahansa syystä (leikkaus, infektio yms): aina hematologin tai PNH-pkl:n konsultaatio (huom. riskitiedot)
- Arvioidaan tarve ekulitsumabihoidon aloitukseen tai ylimääräiseen/aikaistettuun ekulitsumabiannokseen
- Arvioidaan lääkkeellisen tromboosiprofylaksin tarve ellei potilaalla ole pysyvää antikoagulaatiohoitoa.
Kliininen kuva
Kliininen kuva
Yleistä:
PNH:n taudinkuvaan kuuluu klassisesti triadi: krooninen suonensisäinen hemolyysi, verisuonitukostaipumus ja vaikeusasteeltaan vaihteleva luuytimen toimintahäiriö. PNH:n oireet ja kliininen taudinkuva vaihtelevat suuresti: osa potilaista on oireettomia ja toisilla oireet ovat vaikeasti arkielämää ja toimintakykyä rajoittavia. Keskimääräinen elinikä diagnoosista on noin 20 vuotta, mutta vaihtelee paljon (8).
- Krooninen suonensisäinen hemolyysi
- Hemolyysin voimakkuus on yhteydessä PNH-kloonin kokoon sekä komplementin aktiivisuuteen.
- osa potilaista on kroonisesta hemolyysistä huolimatta oireettomia, koska luuytimen hematopoieesi kompensoi hemolyysin
- Hemolyysiin liittyvät oireet:
- yleinen uupumus (fatigue)
- päänsärky
- sileän lihaksen tonuksen toimintahäiriö (hemolyysissä vapautunut Hb sitoo typpioksidia, jonka puutteen oletetaan aiheuttavan oireet)
- vatsa- ja/tai selkäkipu
- pahoinvointi
- nielemiskipu tai – vaikeus
- erektiohäiriö
- Typpioksidin vajaus voi aiheuttaa myös pulmonaalihypertension
- rasitushengenahdistus, heikentynyt rasituksensieto, synkope
- Hemolyysi kiihtyy mm. infektioiden, rokotusten, leikkausten ja vammojen yhteydessä
- Hemolyyttisen kriisin yhteydessä voi ilmaantua akuutti, usein korjaantuva munuaisten vajaatoiminta. Pitkäaikaisseurannassa jopa 64%:lla potilaista on todettu jonkinasteinen munuaisten vajaatoiminta (9).
- Laboratoriolöydökset:
- hemolyysi:
- negatiivinen suora Coombs
- retikulosytoosi
- koholla olevat LD ja bilirubiini (konjugoimaton)
- matala haptoglobiini
- anemia
- hemoglobinuria
- Hb:sta vapautunut rauta hilseilee munuaisen tubulussolujen mukana virtsaan hemosideriininä ja voimakkaan hemolyysin yhteydessä myös Hb pääsee virtsaan tehden virtsasta tummaa, lähes mustaa. (U-Hb, U-hemosideriini +)
- hemolyysi:
2. Verisuonitukostaipumus
- Tukoksia esiintyy eri tutkimusten mukaan 29-44%:lla potilaista (10)
- Tukoksia voi olla sekä valtimoissa että laskimoissa
- Tukoksia voi olla myös epätyypillisissä paikoissa: vatsan alueen laskimoissa,
maksalaskimoissa (Budd-Chiari oireyhtymä) ja aivojen laskimoissa
- Suuri tautiklooni ja voimakas hemolyysi lisäävät tukosriskiä.
- Laskimotukokset ovat tavallisin kuolinsyy PNH-potilailla (8)
3. Luuytimen toimintahäiriö
- Toimintahäiriön aste vaihtelee
- Osalla potilaista tauti ilmenee pääasiassa sytopenioina
- Taudinkuva voi muuttua ajan myötä aplastiseksi anemiaksi tai MDS:ksi, toisaalta
osa potilaista voi parantua spontaanisti.
Luokittelu
Luokittelu
PNH luokittelu taudinkuvan ja löydösten perusteella (1):
Klassinen PNH
- Suonensisäinen hemolyysi: retikulosytoosi, koholla olevat LD ja bilirubiini, matala haptoglobiini
- Ei viitettä luuytimen klonaalisesta sairaudesta
PNH liittyneenä klonaaliseen verisairauteen
- Tavallisimmin aplastinen anemia, myelodysplastinen oireyhtymä
- Muut myeloiset sairaudet
Subkliininen PNH
- Potilaalla voidaan osoittaa PNH-klooni, mutta ei laboratoriokokein
osoitettavaa hemolyysiä
PNH-potilaiden seuranta
PNH-potilaiden seuranta
Potilaan seurannassa pyritään arvioimaan paitsi PNH-taudin aktiivisuutta myös mahdollisia komplikaatioita ja niiden riskiä.
PNH potilaiden seuranta 3-12kk välein:
|
LAB 3-12 kk välein |
MUU Kliinisen harkinnan mukaan |
|
TVK, retik K, Na, Krea, GFRe ALAT, AFOS, Bil LD haptoglobiini APTT, TT/INR, trombai, fibr, FVIII, FIDD ferritiini PNH-klooni 6-12kk välein |
EKG THXrtg Vatsan ultraääni Sydämen ultraääni Punasolusiirtojen tarve |
Jos PNH-kloonin koko <1%, suositellaan kontrollia ainakin kertaalleen 6-12kk kuluttua. Jos klooni hävinnyt tai tilanne stabiili ja potilas oireeton, seuranta voidaan lopettaa.
Ekulitsumabihoitoa saavat:
|
LAB Alkuun 2-4 vko välein, sitten 3-6kk välein |
MUU Aloitettaessa ja harkiten 1-2 kertaa vuodessa |
|
TVK, retik K, Na, Krea, GFRe ALAT, AFOS, Bil LD haptoglobiini APTT, TT/INR, trombai, fibr, FVIII, FIDD ferritiini PNH-klooni 6-12kk välein |
EKG THXrtg Vatsan ultraääni Sydämen ultraääni Punasolusiirtojen tarve Elämänlaatukysely (SF-36) |
- Näytteenotto kannattaa ajoittaa juuri ennen ekulitsumabin antoa (minimivaikutus)
- LD laskee yleensä jo 1-2 infuusion jälkeen
- PNH-kloonin lisääntyminen punasoluissa on merkki tehoavasta hoidosta koska hemolyysi vähenee
- FiDD laskee
PNH-rekisteri
PNH-rekisteri
Kansainväliseen PNH-rekisteriin kerätään potilaan kirjallisella suostumuksella tietoa PNH-potilaista. Tarkoituksena on saada epidemiologista tietoa taudin luonnollisesta kulusta. Rekisteri on avoinna kaikille PNH-tautia sairastaville potilaille hoidosta riippumatta. Alexion Pharmaceuticals tukee rekisteriä taloudellisesti, ja sitä monitoroi ICON clinical research. Lisäksi potilaat kutsutaan mukaan myös Suomen hematologiseen rekisteriin (FHRB).
(PNH-rekisterin kansallinen koordinaattori: anna-elina.armstrong(at)hus.fi)
Muiden maiden suosituksia
- Kirjaudu tai rekisteröidy kommentoidaksesi
Muiden maiden suosituksia
| Liite | Koko |
|---|---|
| 379.81 KB | |
| 125.31 KB | |
| 193.56 KB | |
| 190.04 KB | |
| 116.5 KB | |
| 78.79 KB | |
| 125.31 KB |
Viitteet ja kirjallisuutta
Viitteet ja kirjallisuutta
(1) Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 2005 Dec 1;106(12):3699-3709.
(2) Hill A. The incidence and prevalence of paroxysmal nocturnal hemoglobinuria (PNH) and survival of patients in Yorkshire. Blood 2006;108a:Abstract 985.
(3) Pillemer L, Blum L, Lepow IH, Ross OA, Todd EW, Wardlaw AC. The properdin system and immunity. I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science 1954 Aug 20;120(3112):279-285.
(4) Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Deficiency of an erythrocyte membrane protein with complement regulatory activity in paroxysmal nocturnal hemoglobinuria. Proc Natl Acad Sci U S A 1983 Sep;80(17):5430-5434.
(5) Holguin MH, Fredrick LR, Bernshaw NJ, Wilcox LA, Parker CJ. Isolation and characterization of a membrane protein from normal human erythrocytes that inhibits reactive lysis of the erythrocytes of paroxysmal nocturnal hemoglobinuria. J Clin Invest 1989 Jul;84(1):7-17.
(6) Nicholson-Weller A, March JP, Rosenfeld SI, Austen KF. Affected erythrocytes of patients with paroxysmal nocturnal hemoglobinuria are deficient in the complement regulatory protein, decay accelerating factor. Proc Natl Acad Sci U S A 1983 Aug;80(16):5066-5070.
(7) Takeda J, Miyata T, Kawagoe K, Iida Y, Endo Y, Fujita T, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell 1993 May 21;73(4):703-711.
(8) de Latour RP, Mary JY, Salanoubat C, Terriou L, Etienne G, Mohty M, et al. Paroxysmal nocturnal hemoglobinuria: natural history of disease subcategories. Blood 2008 Oct 15;112(8):3099-3106.
(9) Hillmen P et al ; High incidence of progression to chronic renal insufficiency in patients with paroxysmal nocturnal hemoglobinuria. Blood 2007, 110, 3678
(10) Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood 2013 Jun 20;121(25):4985-96; quiz 5105.
(11) Borowitz MJ, Craig FE, Digiuseppe JA, Illingworth AJ, Rosse W, Sutherland DR, et al. Guidelines for the diagnosis and monitoring of paroxysmal nocturnal hemoglobinuria and related disorders by flow cytometry. Cytometry B Clin Cytom 2010 Jul;78(4):211-230.
(12) Hillmen P, Hall C, Marsh JC, Elebute M, Bombara MP, Petro BE, et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med 2004 Feb 5;350(6):552-559.
(13) Hillmen P, Muus P, Duhrsen U, Risitano AM, Schubert J, Luzzatto L, et al. Effect of the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria. Blood 2007 Dec 1;110(12):4123-4128.
(14) Brodsky RA, Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Schubert J, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood 2008 Feb 15;111(4):1840-1847.
(15) Kelly R, Arnold L, Richards S, Hill A, Bomken C, Hanley J, et al. The management of pregnancy in paroxysmal nocturnal haemoglobinuria on long term eculizumab. Br J Haematol 2010 May;149(3):446-450.
(16) Kelly RJ, Hochsmann B, Szer J, Kulasekararaj A, de Guibert S, Roth A, et al. Eculizumab in Pregnant Patients with Paroxysmal Nocturnal Hemoglobinuria. N Engl J Med 2015 Sep 10;373(11):1032-1039.
Kimmo Porkka, Riitta Lassila, Kari Remes, Eeva-Riitta Savolainen (toim.), Veritaudit 4., uudistettu painos, Duodecim 2015
Canadian immunization guide http://healthycanadians.gc.ca/publications/healthy-living-vie-saine/4-ca...
ACIP CDC https://www.cdc.gov/mmwr/preview/mmwrhtml/mm6441a3.htm
Terveyden ja hyvinvoinnin laitos https://www.thl.fi/fi/web/rokottaminen
Suomen PNH-ryhmä
Elina Armstrong (pj), Hanna Jarva, Eva-Stina Korkama (siht), Minna Lehto, Seppo Meri, Tarja-Terttu Pelliniemi, Eira Poikonen, Jonna Salonen, Marjatta Sinisalo sekä Marjaana Säily
Kiitämme osastonylilääkäri, dosentti Veli-Jukka Anttilaa HUSin infektiosairauksien klinikasta arvokkaista kommenteista ja konsultaatioavusta.
Plasmasolutaudit
- Kirjaudu tai rekisteröidy kommentoidaksesi
Plasmasolutaudit
Myelooma
- Kirjaudu tai rekisteröidy kommentoidaksesi
Myelooma
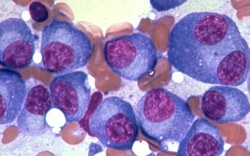 Luokitus: ICD-O 9732/3, ICD-10 C90.0
Luokitus: ICD-O 9732/3, ICD-10 C90.0
Myelooma on pahanlaatuinen verisairaus, plasmasolusyöpä, joka saa alkunsa luuytimen B-solujen malignisoituneesta plasmasolukloonista. Myeloomasolun terve vastinsolu on monenlaisia immunoglobuliineja, vasta-aineita, tuottava B-solulinjan kypsä plasmasolu. Sairastunut plasmasoluklooni tuottaa vain yhdenlaista immunoglobuliinia, jota kutsutaan nimella monoklonaalinen proteiini, M-komponentti tai paraproteiini. Paraproteiini muodostuu yleensä immunoglobuliinin raskas- ja kevytketjusta, mutta noin 15-20% myeloomista erittää pelkästään kevytketjua; kappaa tai lambdaa. IgG-myeloomia on noin 50%, IgA-myeloomia 20%, loput 10% ovat harvinaisia IgD- tai non-sekretorisia myeloomia tai plasmasoluleukemioita. Kevytketjumyelooma voi johtaa munuaisten vajaatoimintaan ja korkea M-komponentti veressä voi aiheuttaa hyperviskositeettioireita. Luuytimessä paikallisesti lisääntyvä myeloomasolukko aiheuttaa multippeleita lyyttisiä luustopesäkkeitä (multippeli myelooma), luustokipua ja jopa patologisia murtumia. Myös paikallisia plasmasolukasvaimia, plasmasytoomia, voi esiintyä luuytimen ulkopuolellakin.
Euroopassa todetaan noin 23 000 uutta tapausta vuodessa, Suomessa ilmaantuvuus on 4,7/100 000/vuosi. Myelooma muodostaa 1% kaikista syövistä ja 10-15% hematologisista syövistä. Myeloomapotilas on keskimäärin 68-70 vuotias taudin toteamishetkellä. Myelooman syy ei ole tiedossa, vaikka useita myeloomassa esiintyviä kromosomimuutoksia jo tunnetaan. Kyseessä ei ole periytyvä sairaus, mutta joissain harvoissa tapauksissa saman perheen ja suvun jäseniltä löytyy erilaisia B-solusarjan pahanlaatuisia veritauteja (Waldenströmin tauti, myelooma, KLL).
Myelooman diagnoosi perustuu luuydinlöydökseen ja paraproteiinin osoittamiseen, mutta myelooman oireisuus ratkaisee hoidon aloituksen.
| Kirjoittajat: | Raija Silvennoinen, Mervi Putkonen, Tarja-Terttu Pelliniemi, Marjaana Säily, Pekka Anttila, Kari Remes, Pia Niittymäki SLR:n myeloomaryhmä |
| © Suomen Hematologiyhdistys | Suomen Leukemiaryhmä |
Diagnoosi
Diagnoosi
Myelooman diagnoosi ja hoidonaloituskriteerit ovat uudistuneet syksyllä 2014. Aiemmin vain CRAB-kriteerien mukaan oireiset potilaat katsottiin hoitoa vaativiksi (myelooman aiheuttama hyperkalsemia, munuaisten vajaatoiminta, anemia, luustotauti). Uuteen kriteeristöön on otettu kolme uutta biomarkkeria, joiden täyttyessä riski kahden vuoden aikana taudilla edetä hoitoa vaativaksi myeloomaksi on niin suuri, että hoidon aloitus on aiheellinen. Myös muutamia muita muutoksia on tehty, mm. myelooman diagnoosi ei enää edellytä paraproteiinin löytymistä. Lisäksi CRAB-kriteereitä on täsmennetty, mm. osteoporoosia ei enää lasketa luustotaudin kriteereihin ja kreatiniinin rinnalle on otettu eGFR munuaisfunktion arviossa.
Uudistuneet diagnoosi ja hoidonaloituskriteerit löytyvät kohdasta plasmasolutaudit.
Diagnoosivaiheen tutkimukset, jotka suositellaan tehtäviksi kaikille
Veri- ja virtsatutkimukset:
TVK, ABORhD, CRP, kalium, natrium, kreatiniini, albumiini, S-Ca, S-Ca-ion, fosfaatti, ALAT, AFOS, bilirubiini, uraatti, LD, beeta2mikroglobuliini, PLV, S- ja dU-proteiinifraktiot (elektroforeesit), S- ja dU- immunofiksaatiot, S-vapaat kevytketjut ja näiden suhde (S-IgLc-V), P-IgG, P-IgA, P-IgM.
Muista myös harvinainen IgD-myelooma (P-IgD, immunofiksaatio IgD- vasta-aineilla) ellei paraproteiinia ala löytyä.
Kuvantamistutkimukset:
Matala-annoksinen luuston TT-kuvaus on ensisijainen kuvantamismenetelmä niissä yksiköissä, joissa se on saatavilla. Luuston TT-kuvaus tehdään ilman varjoainetta, joten kreatiniinillä ei merkitystä.
- Poikkeuksena mikäli diagnoosivaiheessa todetaan ekstramedullaarinen eli luuston ulkopuolinen plasmasytooma (luuston plasmasytoomaan liittyvää pehmytkudoskasvua ei lasketa), niin tällöin perinteinen varjoainetehosteinen vartalon TT-kuvaus on kuitenkin ensisijainen, sillä matala-annoksisen luuston TT-kuvauksen erotuskyky ei riitä ekstramedullaaristen plasmasytoomien toteamiseen.
Niissä yksiköissä, joissa matala-annoksinen luuston TT-kuvaus ei saatavilla käytetään ensisijaisesti edelleen perinteisiä luuston natiivikuvia (kallo, lantio, kaula-, rinta- ja lanneranka, pitkät luut). Perinteiset luustokuvat on hyviä pitkien luiden ja kallon osalta, mutta niiden kyky havaita lantion ja selkärangan lyyttiset muutokset on selvästi heikompi kuin TT-menetelmällä.
Mikäli luuydinnäytteiden ja luuston kuvauksen (luuston TT tai perinteisen luustokuvat) jälkeen jää vaikutelma smoldering myeloomassa, niin lantion ja selkärangan MRI on pakollinen, jotta pystytään erottelemaan smoldering myelooma hoitoa vaativasta multippelista myeloomasta (ks. plasmasolutautien diagnoosikriteerit). Magneettikuvaus osoittaa herkästi pehmytkudosmuutokset, selkäydinkompression ja patologisen luuydininfiltraation. Selkäydinkompressio-oireet ovat indikaatio päivystysmagneettikuvaukselle. MRI pystyy lisäksi erottelemaan benignin osteoporoosin (ja benignin osteoporoottisen murtuman) myelooman aiheuttamasta osteoporoosista (jos MRI:n luuydinsignaali on normaali, niin kyseessä on benigni osteoporoosi).
PET-TT ei kuulu myelooman rutiinikuvantamiseen. Sitä voidaan kuitenkin erityistapauksissa harkita, esimerkkinä non-sekretorinen hoitoa vaativa myelooma, jossa muulla menetelmällä luustotauti osoitettu. Huomioitava, että PET tulkintaa ei ole myeloomassa standardoitu, tutkimukseen voi liittyä vääriä positiivisia löydöksiä, kaikki myeloomaleesiot eivät merkkiainetta kerää, eikä PET kuulu myelooman virallisiin vastearviokriteereihin.
Hammaslääkäri:
Hampaiston tarkistus ja saneeraus on tarpeen ennen luustolääkityksen aloitusta, ks. tukihoidot. OPTG.
Luuydintutkimukset:
Luuydin aspiraatin ja luuydinbiopsian morfologia. Plasmasolujen klonaliteetti voidaan osoittaa luuydinbiopsiasta immunohistokemiallisesti. Mikäli luuydinbiopsiaa ei oteta, tulee virtaussytometria tutkia plasmasolujen klonaaliteetin osoittamiseksi. Virtaussytometrian rooli jäännöstautianalytiikassa on kasvava, ja näin ollen virtaussytometrian tutkiminen intensiivihoitoon soveltuvilta on muutenkin suositeltavaa.
Perinteistä kromosomitutkimusta (G-raita-analyysi) ei enää suositella käytettävän (Suomen myeloomaryhmän päätös 5.11.2015) sen vähäisen informatiivisuuden vuoksi.
Sytogeneettiset tutkimukset tehdään interfaasi FISH tutkimuksella CD138 valikoiduista plasmasoluista. Seuraavat muutokset on liitetty huonompaan ennusteeseen: del(17p) (jos kloonin suuruus 60% tai suurempi), t(4;14), t(14;16), t(14;20), 1q ylimäärä ja del(1p32). Sytogenetiikkaa käytetään riskiarvioinnissa, sillä ei ole merkitystä diagnoosin asettamisessa.
Biopankki näytteet (FHRB) tutkimustarkoitukseen voidaan ottaa halukkailta.
Diagnoosivaiheen tutkimukset, jotka suositellaan tehtäviksi niille potilaille (< 70 v), joille suunnitellaan autologista tai allogeenista intensiivihoitoa
Yllä mainittujen tutkimusten lisäksi:
E-Coombs, hepatiitti-, HIV- ja kuppaserologiat, HLAI-tyypitys (allogeeninen intensiivihoito)
crab
crab
C Hyperkalsemia: s-ca >2,75 mmol/l
R Munuaisten vajaatoiminta: s-krea > 177 umol/l
A Anemia: normokrominen, normosytäärinen anemia, Hb <100 g/l tai laskenut 20 g/l alle normaalin viitealueen
B Luustomuutokset: lyyttiset pesäkkeet, vaikea osteopenia tai patologiset murtumat
Luokittelu
- Kirjaudu tai rekisteröidy kommentoidaksesi
Luokittelu
Durie-Salmon
Durie-Salmon
Vaikeusaste I
Myeloomasolukon määrä on alle 0,6 x 1012 solua/m2. Kaikki alla mainitut kriteerit täyttyvät:
- Hb yli 100 g/l
- S-Ca alle 3,0 mmol/l (normaali)
- Luuston röntgenlöydös on normaali tai todetaan vain yksi osteolyyttinen pesäke
- M-komponentin määrä:
- S-IgG pienempi kuin 50 g/l
- S-IgA pienempi kuin 30 g/l
- Virtsassa pienempi kuin 4 g/vrk
Vaikeusaste II
Myeloomasolukon määrä 0,6 – 1,2 x 1012 solua/m2.
Ne myeloomat, jotka eivät sovi vaikeusasteeseen I tai III.
Vaikeusaste III
Myeloomasolukon määrä yli 1,2 x 1012 solua/m2. Myelooma täyttää yhden tai useampia seuraavista kriteereistä:
- Hb alle 85 g/l
- S-Ca yli 3,00 mmol/l
- Laajat lyyttiset luustomuutokset
- Suuri M-komponentti:
- IgG suurempi kuin 70 g/l
- IgA suurempi kuin 50 g/l
- Virtsassa suurempi kuin 12 g/vrk
Ryhmät A ja B
Vaikeusasteet I, II ja III jaetaan ryhmiin A ja B munuaisfunktion perusteella:
A) P-Krea on < 176 μmol/l
B) P-Krea on ≥176 μmol/l
Durie BG, Salmon SE. A clinical staging system for multiple myeloma. Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer. 1975, 36(3):842-54.
IMWG riskiluokitus
- Kirjaudu tai rekisteröidy kommentoidaksesi
IMWG riskiluokitus
| Suuri riski (high-risk) | Standardiriski (Standard-risk) | Pieni riski (Low-risk) | |
| Muuttujat |
|
Muut |
|
| Mediaani OS | 2 vuotta | 7 vuotta | >10 vuotta |
| % potilaista | 20 % | 60 % | 20 % |
Viitteet: Chng WJ, IMWG consensus on risk stratification in multiple myeloma, Leukemia 2014; 28: 269-27
ISS ja R-ISS
ISS ja R-ISS
ISS
International stageing system (ISS) for multiple myeloma - vaikeusasteen kriteerit
Vaikeusaste |
Ennustetekijät |
Mediaanielinaika (kk) |
|
1 |
B2M < 3,5mg/l ja albumiini ≥ 35g/l |
62 |
|
2 |
B2M < 3,5mg/l ja albumiini < 35g/l tai B2M 3,5 – 5,4 mg/l |
44 |
|
3 |
B2M ≥5,5 mg/l |
29 |
Greipp PR, San Miguel J, Durie BG, Crowley JJ, Barlogie B, Blade J, Boccadoro M, Child JA, Avet-Loiseau H, Kyle RA, Lahuerta JJ, Ludwig H, Morgan G, Powles R, Shimizu K, Shustik C, Sonneveld P, Tosi P, Turesson I, Westin J. International staging system for multiple myeloma. J Clin Oncol. 2005 May 20;23(15):3412-20.
Revised ISS (R-ISS)
| Vaikeusaste | Kriteerit | Mediaanielinaika (kk) | |
| R-ISS 1 |
|
Ei saavutettu | |
| R-ISS 2 | Ei R-ISS 1 tai R-ISS 3 | 83 | |
| R-ISS 3 |
|
43 | |
Palumbo A, Avet-Loiseau H, Oliva S, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol. 2015 Sep 10;33(26):2863-9.
Mitattava tauti
- Kirjaudu tai rekisteröidy kommentoidaksesi
Mitattava tauti
Myelooman hoitoa aloitettaessa arvioidaan onko tauti mitattavissa (measurable) ja millaisella mittarilla.
Mitattavissa olevan taudin (measurable) määritelmä (vuokaavio):
*Luuydinaffiisio tulisi mieluiten arvioida luuydinbiopsialla; jos aspiraatissa ja biopsiassa on epäsuhta käytetään näistä korkeampaa arvoa. Virtaussytometriaa ei käytetä plasmasolumäärän arviointiin.
Mikäli tauti ei ole mitattavissa, vastearviona voidaan käyttää vain täydellistä remissiota (CR) tai sitä syvempää vastetta, sekä progressiivista tautia (PD).
Mikäli hoidon alussa seerumissa (tai virtsassa) M-komponentissa on useampi piikki, niin niiden summaa käytetään vasteen arvioimiseen.
Plasmasolutaudit (luokittelu)
Plasmasolutaudit (luokittelu)
Non-IgM monoklonaalinen gammapatia (Non-IgM monoclonal gammopathy of undetermined significance, Non-IgM MGUS)
Kaikkien kolmen kriteerin on täytyttävä:
• Seerumin monoklonaalinen proteiini (non-IgM) < 30 g/l
• Luuytimessä klonaalisia plasmasoluja < 10 %
• Ei merkkejä kohde-elinvaurioista (CRAB), amyloidoosista
IgM monoklonaalinen gammapatia (IgM MGUS)
Kaikkien kolmen kriteerin on täytyttävä:
• Seerumin IgM monoklonaalinen proteiini < 30 g/l
• Luuytimessä lymfoplasmasyyttinen infiltraatio < 10 %
• Ei merkkejä anemiasta, yleisoireista, hyperviskositeetista, lymfadenopatiasta, hepatosplenomegaliasta, tai muusta lymfoproliferatiivisen taudin aiheuttamasta pääte-elinvauriosta
Kevytketjujen monoklonaalinen gammapatia (Light-chain MGUS)
Kaikkien kriteerien on täytyttävä:
- Poikkeava seerumin vapaiden kevytketjujen suhde ja osallisen (involved) kevytketjun määrä yli viitearvojen ylärajan absoluuttisesti
- Ei immunoglobuliinin raskasketjua immunofiksaatiossa
- Ei merkkejä kohde-elinvaurioista (CRAB), amyloidoosista
- Luuytimessä klonaalisia plasmasoluja < 10 %
- Virtsan monoklonaalinen kevytketjueritys < 500 mg/24h
Smoldering myelooma (ei hoitoa vaativa myelooma) (Smoldering multiple myeloma)
Molempien kriteerien täytyttävä:
• Seerumin monoklonaalinen proteiini (IgG tai IgA) ≥ 30 g/l (tai virtsan monoklonaalinen proteiini ≥ 500 mg/24h) ja/tai luuytimessä klonaalisia plasmasoluja 10-60 % JA
• Ei plasmasolusairaudesta johtuvia kohde-elinvaurioita (CRAB), eikä myeloomaa määrittäviä biomarkkereita, eikä amyloidoosia.
Multippeli myelooma (hoitoa vaativa myelooma) (Multiple myeloma)
Molempien kriteerien on täytyttävä:
• Luuytimessä klonaalisia (virtaussytometria tai immunohistokemia) plasmasoluja ≥ 10% TAI biopsialla osoitettu ekstramedullaarinen plasmasytooma
• Plasmasolutaudista johtuva kohde-elinvaurion löydös (CRAB) TAI jokin myeloomaa määrittävä biomarkkeri:
CRAB - kriteerit:
- Hyperkalsemia: seerumin kalsium > 2.75 mmol/l
- Munuaisten vajaatoiminta: seerumin kreatiniini > 177 µmol/l tai eGFR < 40 ml/min
- Anemia (Hb < 100 g/l tai Hb vähintään 20 g/l laboratorion viitearvoja matalampi) (Suomalaiset viitealueet hemoglobiinille; naiset 117 – 155 g/l, miehet 134 – 167 g/l)
- Luustomuutokset: yksi tai useampi lyyttinen pesäke luuston natiivikuvissa tai TT:ssä (jokaisen tulee olla läpimitaltaan vähintään 5 mm) (jos luuytimessä on < 10% plasmasoluja vaaditaan enemmän kuin yksi lyyttinen leesio solitaarin plasmasytooman jossa matala luuydinaffiisio ja multippelin myelooman erotteluksi).
Myeloomaa määrittävät biomarkkerit:
- klonaalisten plasmasolujen osuus luuytimessä ≥60% (jos epäsuhta luuydinaspiraatin ja -biopsian välillä lasketaan näistä korkeampi arvo)
- osallisten/ei-osallisten (involved/uninvolved) seerumin vapaiden kevytketjujen suhde ≥100 (osallisten kevytketjujen oltava vähintään 100 mg/l absoluuttisesti)
- >1 fokaalileesio MRI:ssä (läpimitaltaan vähintään 5 mm)
Solitaarinen plasmasytooma (Solitary plasmacytoma)
Kaikkien neljän kriteerin täytyttävä
• Yksittäinen biopsialla todennettu luun tai pehmytkudoksen kasvain, jossa klonaalisia plasmasoluja
• Luuydinlöydös klonaalisia plasmasoluja < 10%
(solitaari plasmasytooma jaotellaan luuydinlöydöksen perusteella kahteen entiteettiin: solitaariseen plasmasytoomaan jossa ei lainkaan klonaalisia plasmasoluja luuytimessä sekä solitaariseen plasmasytomaan, jossa minimaalinen luuydinaffiisio <10%)
• Normaali luuston natiivikuvaus sekä selkärangan ja lantion magneettikuvaus (tai TT) (paitsi primaari leesio)
• Ei kohde-elinvaurion merkkejä (CRAB)
Plasmasoluleukemia
Jommankumman kriteerin on täytyttävä:
• Veren valkosoluista > 20 % muodostuu plasmasoluista ja /tai
• Veren plasmasolujen absoluuttinen määrä > 2 x 10E9/l
Systeeminen AL – amyloidoosi (Systemic AL amyloidosis)
Kaikkien neljän kriteerin täytyttävä
• Amyloidin kertymiseen liittyvä poikkeava kliininen oire tai löydös, esim. munuaisten, maksan, sydämen, mahasuolikanavan tai perifeerisen hermoston taholta
• Kudoksesta amyloidivärjäys (Kongopunavärjäys) positiivinen (esim. rasva-aspiraatti, ihon stanssibiopsia, luuydin tai elinbiopsia)
• Immunohistokemiallinen värjäys tai muu menetelmä, joka osoittaa amyloidin olevan kevytketjuamyloidia
• Osoitus monoklonaalisesta plasmasolusairaudesta; seerumin tai virtsan M-proteiini, poikkeava seerumin vapaiden kevytketjujen suhde tai klonaalisia plasmasoluja luuytimessä
POEMS
Diagnoosi edellyttää molempia pakollisia (mandatory) kriteereitä sekä yhtä 3:sta major kriteeristä ja yhtä 6:sta minor kriteeristä.
Pakolliset (mandatory) Major kriteerit (molemmat vaaditaan)
- Polyneuropatia
- Monoklonaalinen plasmasolutauti (lähes aina lambda)
Muut Major kriteerit (1 vaaditaan)
- Castlemanin tauti
- Skleroottiset luustomuutokset
- S/P-VEGF koholla (raja-arvoa ei ole määritetty; IMWG suosittelee että olisi ainakin 3-4 kertaa yli laboratorion viitearvojen ylärajan jotta laskettaisiin major kriteeriksi)
Minor kriteerit (1 vaaditaan)
- Organomegalia (splenomegalia, hepatomegalia, lymfadenopatia)
- Ekstravaskulaarinen nestelasti (turvotus, pleuraneste, askites)
- Endokrinopatia (lisämunuainen, kilpirauhanen, hypofyysi, hypogonadismi, haima, lisäkilpirauhanen); diabetesta tai hypotyreoosia ei lasketa
- Ihomuutokset (hyperpigmentaatio, hypertrichosis, akrosyanoosi, flush, valkoiset kynnet)
- papillaödeema
- Trombosytoosi/polysytemia
Ilmiöillä on oltava ajallinen yhteys, eikä niille saa löytyä muita aiheuttajia. Diagnoosin oikeellisuutta tulee epäillä, mikäli osteoskleroottista pesäkkeitä ei löydy. Sairaudessa nähdään usein myös trombosytoosi ja kohonnut seerumin tai plasman VEGF (vascular endothelial growth factor) – pitoisuus. Suomessa S-VEGF – määritys ei ole kliinisessä rutiinikäytössä, mutta tutkimusta voi tiedustella Göteborgista: Göteborgs Universitet Wallenberglaboratoriet (Göteborg university Wallenberg laboratory for Cardiovascular Research), yhdyshenkilö Associate professor Lillemor Mattsson Hulten, s-posti Lillemor.Mattsson@wlab.gu.se
Laboratoriokeskuksen yleislähete, johon pyynnöksi S - VEGF. Hinta on 340 SEK/näyte.
Viitteet: Rajkumar SV, Dimopoulos MA, Palumbo A et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014 Nov; 15(12): e538-48.
Seuranta ja vastearvio
Seuranta ja vastearvio
- M-komponentti on useimmiten paras taudin aktiviteetin mittari. Induktiohoidon yhteydessä se kannattaa tutkia jokaisen hoidon jälkeen juuri ennen seuraavaa hoitoa.
- Kevytketjumyelooman hoitovasteen mittari on vuorokaudessa erittyvä M-komponentti (dU-prot-fr).
- S-IgLc-V –määritystä voidaan käyttää kevytketjumyelooman seurannassa ja joidenkin non-sekretoristen myeloomien seurannassa.
- Plasman immunoglobuliinitasoja (lähinnä P-IgA, P-IgM ja P-IgD) voidaan käyttää, jos M-komponentti erottuu huonosti proteiinifraktioinnissa.
Taudin status
Taudin status
SHR-muuttuja: status_mm
Standardi IMWG vastearvio kriteerit
|
Lyhenne |
Nimike |
Määritelmä |
|
Dg |
Diagnoosivaihe Diagnosis |
|
|
sCR |
Stringent complete response Täydellinen vaste lisäehdoin |
|
|
CR |
Complete response Täydellinen vaste |
|
|
VGPR |
Very good partial respomse Erittäin hyvä osittainen vaste |
|
|
PR |
Partial response Osittainen vaste |
|
| MR |
Minimal response Minimaalinen vaste |
|
|
SD |
Stable disease Stabiili tauti |
|
|
PD |
Progressive disease Progressiivinen tauti |
TAI
TAI
TAI
|
|
Clinical relapse |
Kliininen relapsi |
|
| Relapse from complete response (CR) |
Relapsi täydellisestä vasteesta (CR) |
|
|
Relapse from MRD |
Relapsi MRD negatiivisuudesta |
|
| Exitus | Exitus |
IMWG MRD kriteerit (edellyttää CR)
| Sustained MRD-negative | Kestävä MRD-negatiivinen |
|
| Flow MRD-negative | Virtaus MRD-negatiivinen |
|
| Sequencing MRD-negative |
Sekvensointi MRD-negatiivinen |
|
| Imaging-positive MRD-negative |
Kuvantamis-vahvistettu MRD-negatiivinen |
|
Huomioitavaa:
Plasmasytoomien koon muutokset arvioidaan SPD perusteella (sum of the products of the maximal perpendicular diameters) (kohtisuorien maksimaalisten läpimittojen tulojen summa).
Sädehoidettua plasmasytoomaa ei voi käyttää vasteen arvioinnissa (paitsi kasvua voidaan käyttää progressiivisen taudin toteamiseksi).
Kaikki vastearviot (paitsi luuydin ja MRD arvio) edellyttävät kahta perättäistä mittausta ennen kuin vaste on varmistettu (paitsi stabiili tauti). Varmistava tutkimus voidaan tehdä milloin vain (myös samana päivänä).
Mikäli hoidon alussa M-komponentti on jakautunut useammaksi piikiksi, niin niiden summaa käytetään vasteen arvioimiseen.
Erityisesti CR saavuttamisen jälkeen voi immunofiksaatiossa ilmetä eri isotyyppiä oleva M-komponentti (joko raskas- tai kevytketju erilainen kuin alunperin). Tämä todennäköisesti kuvaa oligoklonaalista immuunirekonstituutiota, yleensä häviää ajan myötä, eikä sitä pidä sekoittaa relapsiin.
Muita määritelmiä:
| Refraktaari myelooma |
Tauti on refraktaari jos vähintään minimaalista vastetta (MR) ei saavuteta, tai tauti progredioi (PD) hoidon aikana tai 60 vrk sisällä viimeisestä hoidosta. Refraktaari myelooma sisältää 2 kategoriaa:
|
| Hoitolinjan vaihtuminen | Hoitolinja vaihtuu kun uusia lääkkeitä aloitetaan tai lisätään hoitoon progressiivisen taudin (PD) tai kliinisen relapsin jälkeen (Suomen myeloomaryhmän suositus 15.1.2015). |
Viitteet:
Kumar S. International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol. 2016 Aug;17(8):e328-46
Hoito
- Kirjaudu tai rekisteröidy kommentoidaksesi
Hoito
Kansalliset hoitosuositukset (FMG)
Kansalliset hoitosuositukset (FMG)
Suomen myeloomaryhmän (FMG) hoitosuositus
Tärkeimmät muutokset vuoden 2021 hoitosuosituksessa (verrattuna edelliseen 2019 hoitosuosituksen):
- Ensilinjan hoito:
- Autologiseen kantasolusiirtoon soveltuvien induktiohoidon vaihtoehdoista VTD poistettu (VCD induktiohoito jätetty).
- Kantasolusiirtoon soveltumattomien hoitovaihtoehdot ennallaan (VRD, LenDex, VMP), mutta niiden väliseen valintaan vaikuttavia seikkoja tarkennettu.
- Uusiutuneen taudin hoito:
- ASCT jälkeen varhaisen relapsin määritelmä 18 kk siirrosta pidennetty (aiemmin 12 kk).
- Poistettu erillinen kappale kantasolusiirtoon soveltumattomien ensimmäisestä relapsista ja yhdistetty se myöhempiin relapseihin.
- Relapsien hoitovaihtoehtoja on huomattavasti päivitetty ja täsmennetty. Vaihtoehdot on jäsennelty uusiksi.
| Liite | Koko |
|---|---|
| 821.28 KB |
Aiemmat myelooman hoitosuositukset (ei voimassa)
Aiemmat myelooman hoitosuositukset (ei voimassa)
Tälle sivulle on koottu Suomen myeloomaryhmän (FMG) aiempia hoitosuosituksia, jotka eivät ole enää voimassa.
Ensimmäinen yhtenäinen hoitosuositus julkaistiin 2017.
Tärkeimmät muutokset vuoden 2019 hoitosuosituksessa verrattuna edelliseen 2017 hoitosuositukseen:
- Autologisen kantasolusiirron jälkeinen hoito:
- VTD x2 konsolidaatiohoidon tilalle VRD x2 konsolidaatiohoidot.
- Non-HR sytogenetiikka: Lenalidomidi ylläpitohoito progressioon saakka (aiemmin 1 vuosi).
- HR sytogenetiikka: Ylläpitohoitona bortetsomibi + lenalidomidi 1 vuosi, jonka jälkeen lenalidomidi progressioon saakka (aiemmin bortetsomibi 1 vuosi).
- t(14;20) mukaan suuren riskin muutoksiin.
- Kantasolusiirtoon soveltumattomille VMP ja Len/Dex rinnalle VRD hoitovaihtoehdoksi 1. linjassa.
| Liite | Koko |
|---|---|
| 629.97 KB | |
| 747.29 KB |
Tukihoidot myeloomassa
Tukihoidot myeloomassa
Bisfosfonaattihoito
Luustolääkitys (bisfosfonaatti tai denosumabi) tulee aloittaa kaikille myeloomapotilaille, joille aloitetaan ensilinjan myeloomahoito, vaikka ei todettaisikaan lyyttisiä luustomuutoksia. Bisfosfonaattihoitoa (BP) ei tule aloittaa rutiinisti potilailla, joilla on oireeton (smoldering) myelooma tai MGUS. BP voidaan käyttää kuitenkin jos smoldering myelooma tai MGUS potilaalla todetaan osteoporoosi, osteoporoosiin käytettävillä annostuksilla. Jos todetaan solitaari plasmasytooma, mutta ei todeta muuten näyttöä myeloomasta, ei ole tarvetta BP hoitoon.
Bisfosfonaateista tsoledronihappo, pamidronaatti ja klodronaatti ovat hyväksyttyjä EU-alueella. Englantilaisessa tutkimuksessa (MRC-IX) tsoledronihapon todettiin lisäävän jonkin verran elinaikaa ja vähentävän luustotapahtumien riskiä enemmän kuin klodronaatin. Pohjoismaisessa tutkimuksessa todettiin pamidronaatin 30 mg olevan yhtä tehokas kuin 90 mg luustotapahtumien ehkäisyssä. Tsoledronihappo infusoidaan 15 minuutissa ja pamidronaatti 2-4 tunnissa. Tsoledronihapon annosta tulee pienentää jos GFR 30-60 ja pamidronaatin infuusioaikaa pidentää > 4h. Pamidronaattia eikä troledronihappoa suositella, jos GFR < 30. Krea/GFR tulee ottaa aina ennen BP infuusiota.
Denosumabilla on myös indikaatio myelooman luustomuutosten ehkäisyssä ja sitä voidaan käyttää bisfosfonaatin sijaan erityisesti jos GFR on alle 30. Denosumabi annostellaan subkutaanisti.
Ei tiedetä hyödyttääkö bisfosfonaatti tai denosumabin potilaita, joilla luustotautia ei ole todettu nykyaikaisilla kuvantamismenetelmillä (TT, MRI). Aiemmat bisfosfonaattien tutkimukset on tehty aikakaudella ennen nykyaikaisia luuston kuvantamismenetelmiä. Denosumabia ja tsoledronaattia vertailevassa tutkimuksessa (Lancet Oncol. 2018) tutkituilla potilailla oli luustotauti.
Suositus luustotaudin hoidosta ja ennettämisestä (hyväksytty Suomen myeloomaryhmän kokouksessa 28.8.2019):
- Jos GFR≥30, niin bisfosfonaattihoito. Ensisijaisesti tsoledronaatti i.v. 4 viikon välein (GFR mukainen annostelu). Toissijaisesti voidaan käyttää pamidronaatti 30 mg i.v. 4 viikon välein.
- Mikäli mahdollinen munuaisten akuutti vajaatoiminta ei korjaannu siten, että bisfosfonaattia voitaisiin käyttää (GFR jää alle 30): Denosumabi 120 mg s.c. 4 viikon välein.
- Hoidon kesto: IV-bisfosfonaattia käytetään 4 viikon välein 1 vuoden ajan (tai 12 infuusion verran mikäli hoidossa taukoja), jonka jälkeen yleensä siirrytään 3 kk välein annosteluun vielä 1 vuoden ajaksi (hoidon kokonaiskesto 2 vuotta). Denosumabi-hoitoa ei harvenneta. Denosumabin hoidon optimaalisesta kestosta ei tietoa.
- Poikkeuksena ne potilaat, joilla ei ole luustotautia: Jos potilaalla ei ole luustotautia ja saavutaan 1 vuoden kohdalla CR tai VGPR, niin luustolääkitys voidaan lopettaa 1 vuoden hoidon jälkeen..
- Relapsissa luustolääkitys aloitetaan yleensä uudelleen (i.v.-bisfosfonaatti voidaan annostella joko kerran kuukaudessa tai 3 kk välein, riippuen aikaisemman hoidon kestosta).
- Kalsium- ja D-vitamiinilisää suositellaan kaikille aloitettavaksi, kun mahdollinen hyperkalsemia on hoidettu.
Leukaluun osteonekroosi
- Kaikille potilaille hammaslääkärin tutkimus, optimaalisen suuhygienian opettaminen, tarvittavat hammastoimenpiteet.
- Jos hoidon aikana tarvitaan kajoavia hammastoimenpiteitä (hampaan poisto, hammasimplantaatti, leukakirurgia), tulee bisfosfonaattihoito tauottaa 90 ennen ja 90 päivää jälkeen toimenpiteen (IMWG suositus).
- Myeloomaryhmä pitää tärkeänä, että luustolääkityksen aloitus ei viivästyisi hammashoidon takia diagnoosivaiheessa mikäli potilaalla on luustotauti. Tarpeettomia invasiivisia hammastoimenpiteitä tulee välttää ja hammashoito tulisi toteuttaa konservatiivisesti, mikäli mahdollista. Invasiivisen hammastoimenpiteen tarpeellisuus ja ajoitus tulee arvioida hammaslääkärin ja myeloomaa hoitavan lääkärin yhteistyönä. Mikäli aivan akuuttia tarvetta invasiiviselle hammastoimenpiteelle ei ole, niin mahdollinen toimenpide kannattaa lykätä tehtäväksi myöhemmin myeloomatilanteen rauhoituttua. Jos hampaan poisto tarvitsee tehdä pian diagnoosin jälkeen, niin yleensä luustolääkitys kannattaa aloittaa heti kun hampaanpoistoalue on kliinisesti parantunut (esim. 1 kk hampaanpoistosta) eikä odottaa em. 90 päivää.
- Bisfosfonaattia ei tarvitse tauottaa rutiinitoimenpiteitä varten mm. ennen hampaan paikkausta, juurihoitoa tai hammaskiven poistoa.
- Jos todetaan leuan osteonekroosi, tulee bisfosfonaattihoito keskeyttää. Uudelleen aloituksessa tulee punnita yksilökohtaiset hyödyt ja haitat erityisesti myelooman relapsissa tai refraktaarissa taudissa.
Viitteet:
Terpos E. International Myeloma Working Group Recommendations for the Treatment of Multiple Myeloma–Related Bone Disease. Journal of Clinical Oncology 2013;31(18):2347-57 (linkki).
Hyperkalsemian hoito
Hyperkalsemia (Akuuttihoito-opas).
Hyperkalsemia on yleensä merkkinä myelooman aktivoitumisesta. Hyperkalseemisesta kriisistä puhutaan, jos P-Ca on yli 3,75 mmol/l ja P-Ca-Ion on yli 2,0 mmol/l.
Hyperkalsemian hoito
- dehydraation korjaaminen ja diureesin varmistaminen
- nesteytys toteutetaan fysiologisella keittosuolalla
- huolehdi kalium- ja magnesiumtasapainosta
- kalsiumeritystä voidaan tehostaa furosemidilla
- glukokortikoidit; prednisolon tai metyyliprednisolon 30 – 100 mg päivässä
- tsoledronihappo (varovaisuutta munuisten vajaatoiminnassa) tai denosumabi
- kalsitoniini, vaikutus alkaa nopeasti, on lyhytaikainen, soveltuu myös munuaisten vajaatoimintapotilaille
- myelooman spesifinen hoito liitetään mukaan heti, kun se on mahdollista
Kirurginen hoito
Neurokirurginen hoito
- Mikäli myeloomapotilaalla on akuutti parapareesi selkäydinkompressiosta johtuen, konsultoi päivystävää neurokirurgia mahdollisesta leikkauksesta. Dexametasoni 40 mg/ vrk aloitetaan samanaikaisesti.
Vertebroplastia ja kyfoplastia
- Jos nikamaluhistuman aiheuttama selkäkipu ei hellitä konservatiivisin keinoin voidaan selvittää mahdollisuudet vertebroplastiaan tai kyfoplastiaan. Molemmissa tekniikoissa perkutaanisesti injektioidaan luusementtiä nikamaan. Ne ovat tehokkaampia kun ne tehdään lyhyen ajan sisällä nikamaluhistumasta. Molempiin tekniikkoihin liittyy pieni riski sementin vuotamisesta johtaen keuhkoveritulppaan tai neurokompressioon.
Kyriakou et al. The role of cement augmentation with percutaneous vertebroplasty and balloon kyphoplasty for the treatment of vertebral compression fractures in multiple myeloma: a consensus statement from the International Myeloma Working Group (IMWG). Blood Cancer J. 2019 Feb 26;9(3):27
Tromboosiprofylaksia
Myeloomaan liittyy itsellinen laskimotukosriski. Sitä lisäävät vielä infektiot, venakatetrit, obesiteetti, munuaisten vajaatoiminta, ikä.
Immunomodulaattori (IMiD; talidomidi, lenalidomidi, pomalidomidi) hoitoon on liitettävä tromboosiprofylaksia. IMiD monoterapiassa riski on pienempi, mutta yhdistelmähoidossa (deksametasoni, solunsalpaajat) riski nousee noin 10%:iin. Mikäli potilaalla on vain 0-1 riskitekijää, riittää ASA-profylaksia (70-100 mg/vrk). Jos riskitekijöitä on 2 tai useampi tulisi käyttää pienimolekylaarista hepariinia (LMWH) tai varfariinia. Jos varfariini on käytössä muista syistä voidaan sitä jatkaa, mutta LMWH on syöpäpotilaille useimmiten suositeltavampi. Tukosprofylaksiaa tulisi käyttää ainakin ensimmäiset 4-6 kuukautta (tukosriskin ollessa suurin hoidon alussa), jonka jälkeen sen keventämistä voi harkita (esim. siirtyminen LMWH:sta ASA-profylaksiin). Jos tromboosi syntyy, sen hoidossa noudatetaan tavanomaisia hoitokäytäntöjä. Kun tilanne on stabiloitunut immunomodulaattorihoitoa ei yleensä pidetä vasta-aiheisena kunhan antikoagulanttihoito on adekvaatti.
Palumbo A et al. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia 2008:22:414 – 423.
Snowden JA, et al. Guidelines for supportive care in multiple myeloma 2011. BJH 2011:154:76-103
Anemian hoito / Erytropoietiinihoito
Hoidon aiheena on oireinen anemia, yleensä Hb < 100g/l. Vaihtoehtoina eri epoetiinit (alfa, beeta, zeta), jolloin aloitusannos on 30 000- 40 000 KY kerran viikossa sc tai pitkävaikutteinen darbepoetiini 6.25µg/kg 3 vk välein. Jos Hb nousee > 120g/l, EPO-hoito pitää lopettaa/tauottaa.
Sädehoito
Selkäydinkompressiossa on sädehoito aloitettava nopeasti (< 2 vrk), ja hoitoon liitetään korkea-annosdeksametasoni.
Paikallinen sädehoito on usein tehokasta kivunhoitoa kivuliaissa luustopesäkkeissä, annoksena suositellaan 8 Gy kertafraktiona.
Lisäksi sädehoidon indikaationa on plasmasytoomien aiheuttamat kompressio-oireet. Laajoja sädehoitoja luuydinalueille pyritään välttämään ennen kantasolujenkeruuta, jos kliinisesti mahdollista. Hb-tason on oltava sädehoidon aikana > 100 g/l.
Infektiot
- Suositellaan rokotusta influenssaa, Streptococcus pneumoniae (konjugaattirokote) ja Haemophilus influenzae vastaan kaikille, vaikkakin niiden tehokkuus on epävarmaa. Myös perheenjäsenten rokottaminen influenssaa vastaan on suositeltavaa.
- Profylaktista IV-immunoglobuliinihoitoa ei rutiinisti suositella, mutta se saattaa olla hyödyllistä pienellä alaryhmällä potilaita, joilla vaikeita toistuvia bakteeri-infektioita ja hypogammaglobulinemia.
- Profylaktista VZV-profylaksiaa (asikloviiri 400 mg x1) on käytettävä proteasomi-inhibiittori (mm. bortetsomibi) hoitoa saavilla potilailla.
- Kansainvälisissä suosituksissa ei ole otettu kantaa pneumocystis jirovecii estolääkityksen tarpeellisuuteen. Useimmissa kliinisissä myeloomatutkimuksissa pneumocystis estolääkitystä ei ole edellytetty. Suositellaan toimimaan oman keskuksen käytännön mukaisesti.
Palliatiivinen hoito
Tarkoituksena on kivun, taudin aiheuttamien oireiden ja kärsimysten lievittäminen. Tässä vaiheessa ei ole enää tärkeää saada kontrolloitua tautivastetta, vaan potilaan oireita.
Kivun lievitykseen käytetään syöpäpotilaan kipulääkkeitä, ja paikallisia kipusädehoitoja. Vaikeissa tilanteissa on syytä tehdä kipupoliklinikan konsultaatio. Ummetusta ehkäisevä lääkitys, sopiva nesteytys, ja suun kuivamisoireiden hoito on tärkeää. Psyykkisestä tuesta huolehditaan, ja terminaalivaiheen lähestyessä potilaan omat toiveet käydään läpi.
Tukosriskin arviointi immunomoduloivan (IMiD) hoidon yhteydessä
- Kirjaudu tai rekisteröidy kommentoidaksesi
Tukosriskin arviointi immunomoduloivan (IMiD) hoidon yhteydessä
Potilasperäiset ja myeloomasta johtuvat riskitekijät:
- Tuore myelooma
- Hyperviskositeetti
- Aiemmin sairastettu syvälaskimotukos tai suvussa esiintyvä laskimotukosalttius
- Ylipaino (BMI ≥30)
- Komorbiditeetit: sydän, diabetes, munuaisten vajaatoiminta, krooninen tulehduksellinen sairaus
- Immobiliteetti (akuutti tai krooninen)
- Trombofilia, myeloproliferatiivinen tauti, hemoglobinopatia
- Hiljattainen kirurgia (6 viikon sisällä)
- Lääkitykset: erytropoieesia stimuloivat lääkkeet (ESA), hormonikorvaushoito, tamoksifeeni
Jos 0-1 riskitekijää (eikä mitään alla mainituista myeloomaan hoitoon liittyvistä riskitekijöistä): harkitse ASA (70-100 mg päivässä).
Jos 2 tai enemmän riskitekijää: LWMH tai varfariini.
Myelooman hoitoon liittyvät riskitekijät:
- Doksorubisiini
- Suuri-annoksinen steroidi (≥480 mg/kk deksametasonia tai vastaava)
- Kombinaatiokemoterapia
Jos jokin yllä mainituista myelooman hoitoon liittyvistä riskitekijöistä, niin LMWH tai varfariini.
Myelooman hoito yli 70-75 vuotiailla
Myelooman hoito yli 70-75 vuotiailla
Iäkkäiden hoitokuntoisuuden arviointiin ja tutkimusten yhtenäistämiseksi on kehitetty geriatrinen arviointimittari (Myeloma Frailty Score Calculator) (linkki). Sen käyttö tutkimusten ulkopuolella on kuitenkin työlästä. Käytännönläheisempi lähestymistapa on pitää heikkokuntoisena (frail) henkilöä, joka tarvitsee apua kodinhoidossa tai itsestään huolehtimisessa.
Yli 75 vuotiaille, heikkokuntoisille sekä niille joilla on merkittäviä perussairauksia, suositellaan lääkehoitoa annosvähennyksin.
Ks. Suomen myeloomaryhmän (FMG) kansallinen hoitosuositus (alkaen sivu 7): kohta "transplant-ineligible - ensilinjan hoito".
Myeloomahoitojen annosreduktiot
- Kirjaudu tai rekisteröidy kommentoidaksesi
Myeloomahoitojen annosreduktiot
Riskitekijät :
1. Ikä yli 75 v
2. Heikkokuntoisuus (frailty):
- Avuntarve kodinhoidossa tai itsestään huolehtimisessa (pukeutuminen, peseytyminen, WC-käynti, ruokailu)
3. Komorbiditeetit:
- Sydämen toimintahäiriö
- Keuhkojen toimintahäiriö
- Maksan toimintahäiriö
- Munuaisten toimintahäiriö
0 riskitekijää : käytä annostasoa 0 ("Go-Go")
1 riskitekijä : käytä annostasoa -1 ("Moderate-Go")
1 riskitekijä + gradus 3-4 non-hematologinen haittavaikutus (AE) : käytä annostasoa -2 ("Slow-Go")
Viitteet:
Palumbo A, Anderson K. Multiple myeloma. N Engl J Med 2011;364(11):1046-1060.
Palumbo A, Bringen S, Ludwig H, et al. Personalized therapy in multiple myeloma according to patient age and vulnerability: a report of the European Myeloma Network (EMN). Blood 2011; 118(17): 4519-29
Zweegman S, Engelhardt M, Larocca A; EHA SWG on ‘Aging and Hematology’. Elderly patients with multiple myeloma: towards a frailty approach? Curr Opin Oncol. 2017 Sep;29(5):315-321. (referoitu myös: Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up 2021, Suppl)
|
Lääke |
Annostaso 0 |
Annostaso -1 |
Annostaso -2 |
|
Bortetsomibi |
1,3 mg/m2 SC D1, 4, 8, 11 3 vkon sykli |
1,3 mg/m2 SC D1, 8, 15, 22 5 viikon sykli |
1,0 mg/m2 SC D1, 8, 15, 22 5 viikon sykli |
| Karfiltsomibi |
(KRD yhdistelmässä) 20 mg/m2 D1-2, 8-9, 15-16 syklissä 1, 27 mg/m2 syklistä 2 |
(KRD yhdistelmässä) 20 mg/m2 sykli 1 → 27 mg/m2 sykli 2, päivinä 1, 8, 15 |
(KRD yhdistelmässä) 20 mg/m2 päivinä 1, 8, 15 |
| Iksatsomibi |
4 mg PO D1, 8, 15 4 viikon sykli |
3 mg PO D1, 8, 15 4 viikon sykli |
2,3 mg PO D1, 8, 15 4 viikon sykli |
|
Talidomidi |
100 mg/pv PO |
50 mg/pv PO |
50 mg j.t.p PO |
|
Lenalidomidi |
25 mg/pv PO D1-21 4 viikon sykli |
15 mg/pv PO D1-21 4 viikon sykli |
10 mg/pv PO D1-21 4 viikon sykli |
| Pomalidomidi |
4 mg PO D1-21 4 viikon sykli |
3 mg PO D1-21 4 viikon sykli |
2 mg D1-21 4 viikon sykli |
|
Deksametasoni |
40 mg/pv PO D1, 8, 15, 22 4 viikon sykli |
20 mg/pv PO D1, 8, 15, 22 4 viikon sykli |
10 mg/pv PO D1, 8, 15, 22 4 viikon sykli |
|
Melfalaani |
0,25 mg/kg PO (9 mg/m2) D1-4 / 4-6 viikkoa |
0,18 mg/kg PO (7,5 mg/m2) D1-4 / 4-6 viikkoa |
0,13 mg/kg PO (5 mg/m2) D1-4, 4-6 viikkoa |
|
Prednisoni |
60 mg/m2 PO D1-4 TAI 50 mg j.t.p PO |
30 mg/m2 PO D1-4 TAI 25 mg PO j.t.p. |
15 mg/m2 PO D1-4 TAI 12,5 mg PO j.t.p. |
|
Syklofosfamidi |
300 mg/m2 PO D1, 8, 15, 22 TAI 100 mg/pv PO D1-21 4 viikon sykli |
150 mg/m2 PO D1, 8, 15 TAI 50 mg/pv PO D1-21 4 viikon sykli |
75 mg/m2 PO D 1, 8, 15 TAI 50 mg j.t.p. PO D1-21 4 viikon sykli |
Sädehoito
Sädehoito
Selkäydinkompressiossa on sädehoito aloitettava nopeasti (< 2 vrk), ja hoitoon liitetään korkea-annosdeksametasoni.
Muita sädehoidon indikaatioita ovat kivuliaat luustopesäkkeet, patologiset murtumat ja kompressio-oireita aiheuttavat plasmasytoomat.
Laajoja sädehoitoja luuydinalueille pyritään välttämään ennen kantasolujenkeruuta, jos kliinisesti mahdollista.
Hb-tason on oltava sädehoidon aikana > 100 g/l
Kantasolujensiirto
Kantasolujensiirto
Autologisen kantasolujensiirron aiheet
- Toistaiseksi autologinen kantasolujensiirto on standardihoito ensilinjan hoitona <66 (-70v) vuotiailla potilailla, jotka ovat saaneet hyvän vasteen (VGPR- hyvä PR vähintään) induktiohoidolle. Yleinen suositus on 3 - 4 induktiohoitoa ennen kantasolujenkeräystä. Induktiovaiheessa toksisuus kuitenkin lisääntyy, mitä enemmän hoitolinjoja käydään läpi vasteeseen pyrkiessä.
Allogeenisen kantasolujensiirron aiheet
Primaarivaihe
Konsultoidaan allogeenista siirtokeskusta, jos nuori – keski-ikäinen potilas ja
• ei allogeenista siirtoa estäviä muita sairauksia
• suuren riskin taudin piirteitä: suuri tuumoritaakka, aggressiivinen taudinkuva,
• plasmasoluleukemia, ekstramedullaarinen tauti,
• sytogenetiikka: 17p del ja /tai t(4;14), t(14;16), ja erityisesti jos vielä ISS 3
Relapsi/progressiovaihe:
Yleensä sytogenetiikka tutkitaan uudelleen, ja jos on tullut suuren riskin löydöksiä, relapsi käyttäytyy aggressiivisesti ja potilas on allosiirtokuntoinen. Jos relapsi tulee nopeasti, konsultoidaan siirtokeskusta herkästi lievemmissäkin tapauksissa. Allogeeninen siirto ei ole enää järkevä hoito-optio, kun potilaalla on ollut jo useita taudin uusiutumisia.
Autologisen kantasolusiirron toteutus
Autologisen kantasolusiirron toteutus
Autologisen kantasolusiirteen keräys
Autologiset kantasolut kerätään induktiohoidon jälkeen. Induktiohoito tauotetaan 2 – 4 vk ennen kantasolujen keräystä.
Standardi mobilisaatiohoito on syklofosfamidi 2 g/m2 + filgrastiimi 5 – 10 µg/kg/pv tai pegyloitu filgrastiimi 6 – 12 mg. Jos saalis on riittämätön, mikä on harvinaista (paitsi, jos taustalla on laaja sädehoito + talidomidi + runsaasti muita hoitoja), suositellaan uutta keräysyritystä yhdistelmällä: filgrastiimi +/- plerixafor.
Keräyssaaliin minimitavoite on 2 x 10E6 CD34+ solua/kg yhteen siirtoon, mutta suositeltava saalistavoite on 4 x 10E6 solua/kg /siirto. Mikäli mahdollista, pyritään keräämään 8 – 10 x 106 solua/kg, mikä mahdollistaa kaksi täysimittaista autologista intensiivihoitoa (IMWG - konsensus). Toisen intensiivihoidon asema ei ole enää yhtä selvä kuin oli ennen myelooman uusien lääkkeiden (ns.”novel agents”) aikakautta.
Tavoitteena on saavuttaa hoidoin vähintään PR - vaste ennen kantasolujen mobilisaatiota. Joskus voidaan harkinnan mukaan tyytyä siihen, että on saavutettu vain stabiili tautivaste. Tarvittaessa hoitovastetta voidaan pyrkiä syventämään ennen intensiivihoitoa jatkamalla talidomidi + deksametasonihoitoa pitempään. Jos vaste tälle hoidolle jää puutteelliseksi, toisen linjan hoitona käytetään useimmiten bortetsomibi + deksametasonihoitoa (korkea-annosdeksametasoni). Jos tauti progredioi talidomidi + deksametasonihoidon aikana, voidaan harkita siirtymistä suoraan kolmen lääkkeen yhdistelmähoitoon, kuten esim. PAD - tai CVD - hoitoon. Kolmannen linjan hoitona voi tarvittaessa käyttää lenalidomidi + dexametasonihoitoa (matala-annosdeksametasoni, 40 mg x 1 /vk + tromboosiprofylaksia). Jos myelooma on primaaristi refraktääri, käytetään 3. - 4. linjan hoitovaihtoehtoja: VDT-PACE, RVD, RCD, DT-PACE, DCEP, HyperCVAD.
Autologinen intensiivihoito ja kantasolujen palautus
Esihoitona melfalaani 200 mg/m2 (hemodialyysipotilaat/munuaisfunktio alentunut: maksimiannos 140 mg/m2)
Filgrastiimi- tuki siirron jälkeen harkinnan mukaan.
Siirteen tarttumisen päivä = kun B-neutrofiilit > 0,5 x 109/l, kirjataan.
Siirteen tarttumisen varmistaminen: neutrofiilien pysyttävä em. tasolla kolme vrk.
Paras hoitovaste arvioidaan 3 kk kohdalla siirrosta.
Jos 1. siirron vaste on huonompi kuin VGPR, voidaan harkita tehtäväksi tandem-siirto 4 – 6 kk kuluttua 1. siirrosta.
Intensiivihoidolla saavutetussa VGPR/CR – tilanteessa ei käytetä ylläpitohoitoa. PR - tilanteessa voi harkita konsolidaatiohoitoa/lisähoitoja hoitovasteen syventämiseksi. Rutiininomaisen konsolidaatiohoidon puolesta ei ole kuitenkaan vielä riittävää näyttöä. Mikäli kyseessä on huonon ennusteen tauti, eikä allogeenisen siirron mahdollisuutta ole, konsolidaatio – ja ylläpitohoitoa on harkittava (bortetsomibi, lenalidomidi).
Rokotussuoja rakennetaan uudelleen 6 kk kuluttua siirrosta, alueellisten käytäntöjen mukaan.
Autologisen intensiivihoidon jälkeinen seuranta
VGPR/CR - vasteen saavuttaneet
Seuranta ja laboratoriokokeet (PVK, krea, ionisoitunut Ca, AFOS, CRP, LD, S- ja dU-proteiinifraktiot) 2 – 4 kk välein, harventaen edelleen, jos hyvä vaste säilyy. CR- vasteen varmistaminen edellyttää vähintään kerran vuodessa tapahtuvaa S- ja dU- immunofiksaatioiden tarkistamista S- ja dU- proteiinifraktiointien lisäksi, sekä luuydinnäytteen tutkimista. Non-sekretoorisessa myeloomassa tutkitaan em. lisäksi S-vapaat kevytketjut. Stringent CR – vasteen varmistamisessa käytetään edellä mainittujen lisäksi virtaussytometristä jäännöstautimääritystä.
PR – vasteen saavuttaneet
Joissakin tapauksissa, kuten PR – vasteessa, totaali-immunoglobuliinitason (S- IgG, S-IgA) seuraaminen voi riittää. Se ei kuitenkaan kerro tarkkaa paraproteiinin määrää, ja progressio-epäilyssä ja/tai vasteen parantuessa on tarkistettava S- ja dU- proteiinifraktioinnit ja – immunofiksaatiot.
Aiemmat FMG tutkimukset
Aiemmat FMG tutkimukset
Suomen myeloomaryhmän (FMG) aiemmista kliinisistä tutkimuksista löydät tietoa täältä. Kyseiset tutkimukset eivät enää rekrytoi.
Information on former clinical studies organized by the Finnish Myeloma Group can be found here
FMGMM01 (Veldex)
FMGMM01 (Veldex)
VelDex tutkimuksen aktiivivaihe on päättynyt, ja artikkeli julkaistu BJH Feb 2013. Kiitos kaikille tutkimukseen osallistuneille yhteistyöstä! Molekulaarisessa remissiossa olevilta pyydän vielä ottamaan toistaiseksi PCR-seurantanäytteitä, koska apurahaa on jäljellä. Muiden potilaiden PFS/OS aikaa seurataan.
Tutkimuksen arvioitu seurantavaiheen päättymisaika on 31.12.2014.
Kliiniset tutkimukset (myelooma)
Kliiniset tutkimukset (myelooma)
Tietoa kliinisistä myeloomatutkimuksista Suomessa.
Helsingin ja Uudenmaan sairaanhoitopiirissä (HUS) menossa olevat kliiniset tutkimukset löytyvät HUS:n kotisivuilta (linkki).
TYKS-Erva alueen tutkimukset löytyvät Läntisen Syöpäkeskuksen kotisivuilta (linkki).
TAYS Syöpäkeskuksen tutkimukset löytyvät TAYS:n kotisivuilta (linkki).
OYS ja Pohjois-Pohjanmaan sairaanhoitopiirin (PPSHP) tutkimukset löytyvät Pohjoisen Syöpäkeskuksen kotivuilta (linkki).
Pohjoismainen myeloomaryhmän (NMSG) tutkimukset löytyvät NMSG:n kotisivuilta (linkki).
Seuraavat tutkimukset eivät enää rekrytoi uusia potilaita:
- NMSG#23/15. Faasin 2 tutkimus iksatsomibi + lenalidomidi + deksametasonin (IRd) vaikutuksesta jäännöstautiin tuoreessa myeloomassa kantasolusiirtoon soveltuvilla (yhteenvetoon linkki). Rekrytointi päättynyt 3/2020.
- NMSG#20/13. Faasin 2 tutkimus karfiltsomibin käytöstä induktio-, korkea-annos- ja ylläpitohoidossa järjestyksessään toisen autologisen kantasolusiirron yhteydessä potilailla, joiden tauti etenee ensimmäisen siirron jälkeen (linkki). Rekrytointi päättynyt 4/2018.
- NMSG#24/15. Faasin 2 tutkimus karfiltsomibin + elotuzumabin + deksametasonin käytöstä 1-3 aiemman hoitolinjan jälkeen uusiutuneen tai etenevän myelooman hoidossa (protokollaan linkki). Rekrytointi päättynyt 2/2019.
FMGMM02
- Kirjaudu tai rekisteröidy kommentoidaksesi
FMGMM02
Uusien potilaiden rekrytointi päättynyt.
FMGMM02-tutkimuksen protokolla ja 1. amendmentti on ladattavissa alla olevista liitteistä.
The final version and the 1st amendment to the FMGMM002 study protocol can be downloaded from the links at the bottom of the page.
| Liite | Koko |
|---|---|
| 2.02 MB | |
| 237.33 KB |
Potilasohje
- Kirjaudu tai rekisteröidy kommentoidaksesi
Potilasohje
| Liite | Koko |
|---|---|
| 429.16 KB |
Systeeminen AL-amyloidoosi
- Kirjaudu tai rekisteröidy kommentoidaksesi
Systeeminen AL-amyloidoosi
Otsikko vaihdettiin muodosta <em class="placeholder">Systeeminen AL-amyloidoosi</em> muotoon <em class="placeholder">Systeeminen AL-amyloidoosi</em>.
Systeeminen AL-amyloidoosi
- Kirjaudu tai rekisteröidy kommentoidaksesi
Systeeminen AL-amyloidoosi
Systeemisen AL-amyloidoosin hoito-ohje löytyy täältä (PDF).
| Liite | Koko |
|---|---|
| 333.92 KB | |
| 377.71 KB |
Aiemmat AL-amyloidoosin hoitosuositukset (ei voimassa)
Aiemmat AL-amyloidoosin hoitosuositukset (ei voimassa)
Tälle sivulle on koottu Suomen myeloomaryhmän (FMG) aiempia AL-amyloidoosin hoitosuosituksia, jotka eivät ole enää voimassa.
Ensimmäinen yhtenäinen hoitosuositus julkaistiin 2019.
| Liite | Koko |
|---|---|
| 565.21 KB |
Taudin status
- Kirjaudu tai rekisteröidy kommentoidaksesi
Taudin status
SHR-muuttuja: status_amyloid_H (hematologinen vaste), status_amyloid_O (amyloidoosin elinvaste)
Systeemisessä AL-amyloidoosissa arvioidaan erikseen hematologinen vaste, sekä elinvaste.
Kansainvälisesti hyväksytyt elinvastekriteerit olemassa vain sydämen ja munuaisten osalta (tilanne 30.1.2016).
Lähtötason seerumin kevytketjuerotus (dFLC) tulee olla ≥50 mg/l, jotta hematologista vastetta voitaisiin arvioida.
| Hematologinen vaste | |||
|---|---|---|---|
| Koodi | Lyhenne | Nimike | Määritelmä |
| 1 | Dg |
Diagnosis Diagnoosi |
|
| 2 | CR |
Complete response Täydellinen vaste |
Negatiivinen S- ja dU-ImmFix ja normaali seerumin kevytketjujen κ/λ suhde |
| 3 | VGPR |
Very good partial response Erittäin hyvä osittainen vaste |
Osallisten ja ei-osallisten seerumin kevytketjujen erotus alle 40 mg/l |
| 4 | PR |
Partial response Osittainen vaste |
Osallisten ja ei-osallisten seerumin kevytketjujen erotuksen lasku >50% lähtötasosta |
| 5 | NR |
No response Ei vastetta |
PR kriteeri ei täyty |
| Kardiaalinen vaste | ||
|---|---|---|
| Koodi | Nimike | Määritelmä |
| 1 |
Cardiac response Kardiaalinen vaste |
|
| 2 |
Cardiac progression Kardiaalinen progressio |
|
* Koska NT-proBNP eliminaatio riippuu lähes täydellisesti glomerulus filtraatiosta, niin NT-proBNP:tä ei tulisi käyttää kardiaalisen vasteen tai progression merkkinä, niillä joilla munuaisten vajaatoiminta etenee.
| Renaalinen vaste | ||
|---|---|---|
| Koodi | Nimike | Määritelmä |
| 11 |
Renal response Renaalinen vaste |
|
| 12 |
Renal progression Renaalinen progressio |
|
Viitteet:
- Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol 2012;30(36):4541-4549
- Palladini G, Hegenbart U, Milani P, et al. A staging system for renal outcome and early markers of renal response to chemotherapy in AL amyloidosis. Blood 2014 Oct.
- Comenzo RL, Reece D, Palladini G, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis, Leukemia 2012 Nov; 26(11): 2317-25.
Waldenströmin tauti
Waldenströmin tauti
Waldenströmin taudin hoito-ohje löytyy täältä (PDF).
| Liite | Koko |
|---|---|
| 423.98 KB | |
| 406.25 KB |